
Stretching of BDT-gold molecular junctions: thiol or thiolate termination?
Abstract
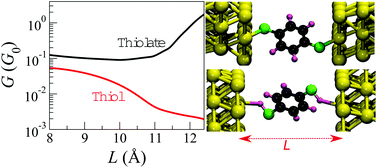
It is often assumed that the hydrogen atoms in the thiol groups of a benzene-1,4-dithiol dissociate when Au–benzene-1,4-dithiol–Au junctions are formed. We demonstrate, by stability and transport property calculations, that this assumption cannot be made. We show that the dissociative adsorption of methanethiol and benzene-1,4-dithiol molecules on a flat Au(111) surface is energetically unfavorable and that the activation barrier for this reaction is as high as 1 eV. For the molecule in the junction, our results show, for all electrode geometries studied, that the thiol junctions are energetically more stable than their thiolate counterparts. Due to the fact that density functional theory (DFT) within the local density approximation (LDA) underestimates the energy difference between the lowest unoccupied molecular orbital and the highest occupied molecular orbital by several electron-volts, and that it does not capture the renormalization of the energy levels due to the image charge effect, the conductance of the Au–benzene-1,4-dithiol–Au junctions is overestimated. After taking into account corrections due to image charge effects by means of constrained-DFT calculations and electrostatic classical models, we apply a scissor operator to correct the DFT energy level positions, and calculate the transport properties of the thiol and thiolate molecular junctions as a function of the electrode separation. For the thiol junctions, we show that the conductance decreases as the electrode separation increases, whereas the opposite trend is found for the thiolate junctions. Both behaviors have been observed in experiments, therefore pointing to the possible coexistence of both thiol and thiolate junctions. Moreover, the corrected conductance values, for both thiol and thiolate, are up to two orders of magnitude smaller than those calculated with DFT-LDA. This brings the theoretical results in quantitatively good agreement with experimental data.
 Please wait while we load your content...
Please wait while we load your content...

