
Density functional theory calculations of catalytic mechanistic pathways for the formation of O2 involving triazolylidene iridium complexes†
Abstract
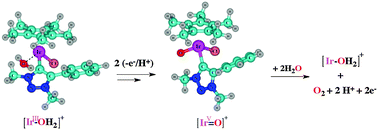
We have studied Ir(III)-catalysed water oxidation pathways at the CAM-B3LYP level of Density Functional Theory (DFT), involving a novel oxygen-evolving catalyst (OEC), for a low-concentration mechanism. Starting from the dichloride complex IrIII–Cl (Ir = [IrCp*Cl(triazolylidene)], Cp* = C5Me5−), DFT predicts reaction with one water molecule and formation of a cationic solvento complex [IrIII–OH2]+, followed by two proton-coupled electron transfer (PCET) steps; one involves an [IrIII–OH2]+/[IrIV–OH]+ transition, whilst the other is an [IrIV–OH]+ to [IrV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]+ transformation. The oxidation potentials and pKa values were calculated for the transformation of [IrIII–OH2]+ to [IrVO]+ for both sequential orders of H+/e− and e−/H+ transfer. Deprotonation of [IrIII–OH2]+ was found to be feasible even in acidic conditions (with a pKa of 1.08), while the deprotonation of [IrIV–OH]+ is strongly disfavoured (pKa = 10.33). The computed oxidation potentials are rather high (1.81 V and 2.01 V for [IrIII–OH2]+ and [IrVI–OH]+, respectively); however, subsequent proton dissociation is more favoured (with pKa values for the oxidised intermediates of −16.1 and −8.1, respectively). This indicates the possibility of concerted, as opposed to stepwise, H+/e− transfer (PCET). Further, the [IrVO]+ intermediate was found to react with two water molecules to release O2. This sequence involves O–O bond formation followed by one PCET step, while a further single electron transfer (oxidation) step generates an [IrV–O–O]+ species, which liberates O2 and regenerates the [IrIII–OH2]+ complex.
O]+ transformation. The oxidation potentials and pKa values were calculated for the transformation of [IrIII–OH2]+ to [IrVO]+ for both sequential orders of H+/e− and e−/H+ transfer. Deprotonation of [IrIII–OH2]+ was found to be feasible even in acidic conditions (with a pKa of 1.08), while the deprotonation of [IrIV–OH]+ is strongly disfavoured (pKa = 10.33). The computed oxidation potentials are rather high (1.81 V and 2.01 V for [IrIII–OH2]+ and [IrVI–OH]+, respectively); however, subsequent proton dissociation is more favoured (with pKa values for the oxidised intermediates of −16.1 and −8.1, respectively). This indicates the possibility of concerted, as opposed to stepwise, H+/e− transfer (PCET). Further, the [IrVO]+ intermediate was found to react with two water molecules to release O2. This sequence involves O–O bond formation followed by one PCET step, while a further single electron transfer (oxidation) step generates an [IrV–O–O]+ species, which liberates O2 and regenerates the [IrIII–OH2]+ complex.
 Please wait while we load your content...
Please wait while we load your content...

