
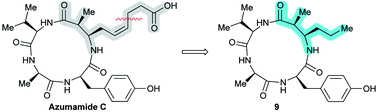
An azumamide C analogue without the zinc-binding functionality†
Abstract
Histone deacetylase (HDAC) inhibitors have attracted considerable attention due to their promise as therapeutic agents. Most HDAC inhibitors adhere to a general “cap-linker-Zn2+-binding group” architecture but recent studies have indicated that potent inhibition may be achieved without a Zn2+-coordinating moiety. Herein, we describe the synthesis of an azumamide analogue lacking its native Zn2+-binding group and evaluation of its inhibitory activity against recombinant human HDAC1–11. Furthermore, kinetic investigation of the inhibitory mechanism of both parent natural product and synthetic analogue against HDAC3-NCoR2 is reported as well as their activity against Burkitt's lymphoma cell proliferation.
 Please wait while we load your content...
Please wait while we load your content...