
From charge transport parameters to charge mobility in organic semiconductors through multiscale simulation
Abstract
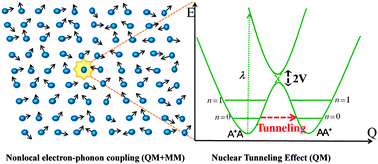
This review introduces the development and application of a multiscale approach to assess the charge mobility for organic semiconductors, which combines quantum chemistry, Kinetic Monte Carlo (KMC), and molecular dynamics (MD) simulations. This approach is especially applicable in describing a large class of organic semiconductors with intermolecular electronic coupling (V) much less than intramolecular charge reorganization energy (λ), a situation where the band description fails obviously. The charge transport is modeled as successive charge hopping from one molecule to another. We highlight the quantum nuclear tunneling effect in the charge transfer, beyond the semiclassical Marcus theory. Such an effect is essential for interpreting the “paradoxical” experimental finding that optical measurement indicated “local charge” while electrical measurement indicated “bandlike”. Coupled MD and KMC simulations demonstrated that the dynamic disorder caused by intermolecular vibration has negligible effect on the carrier mobility. We further apply the approach for molecular design of n-type materials and for rationalization of experimental results. The charge reorganization energy is analyzed through decomposition into internal coordinates relaxation, so that chemical structure contributions to the intramolecular electron–phonon interaction are revealed and give helpful indication to reduce the charge reorganization energy.
 Please wait while we load your content...
Please wait while we load your content...

