
Ab initio Kinetic Monte Carlo simulations of dissolution at the NaCl–water interface†
Abstract
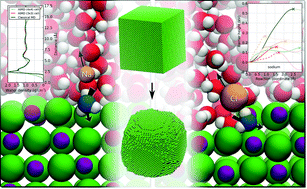
We have used ab initio molecular dynamics (AIMD) simulations to study the interaction of water with the NaCl surface. As expected, we find that water forms several ordered hydration layers, with the first hydration layer having water molecules aligned so that oxygen atoms are on average situated above Na sites. In an attempt to understand the dissolution of NaCl in water, we have then combined AIMD with constrained barrier searches, to calculate the dissolution energetics of Na+ and Cl− ions from terraces, steps, corners and kinks of the (100) surface. We find that the barrier heights show a systematic reduction from the most stable flat terrace sites, through steps to the smallest barriers for corner and kink sites. Generally, the barriers for removal of Na+ ions are slightly lower than for Cl− ions. Finally, we use our calculated barriers in a Kinetic Monte Carlo as a first order model of the dissolution process.
 Please wait while we load your content...
Please wait while we load your content...

