
Ultrafast resonance energy transfer in the umbelliferone–alizarin bichromophore†
Abstract
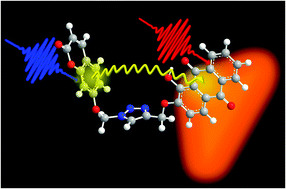
In this work we present the synthesis, time-resolved spectroscopic characterization and computational analysis of a bichromophore composed of two very well-known naturally occurring dyes: 7-hydroxycoumarin (umbelliferone) and 1,2-dihydroxyanthraquinone (alizarin). The umbelliferone donor (Dn) and alizarin acceptor (Ac) moieties are linked to a triazole ring via σ bonds, providing a flexible structure. By measuring the fluorescence quantum yields and the ultrafast transient absorption spectra we demonstrate the high efficiency (∼85%) and the fast nature (∼1.5 ps) of the energy transfer in this compound. Quantum chemical calculations, within the density functional theory (DFT) approach, are used to characterize the electronic structure of the bichromophore (Bi) in the ground and excited states. We simulate the absorption and fluorescence spectra using the TD-DFT methods and the vertical gradient approach (VG), and include the solvent effects by adopting the conductor-like polarizable continuum model (CPCM). The calculated electronic structure suggests the occurrence of weak interactions between the electron densities of Dn and Ac in the excited state, indicating that the Förster-type transfer is the appropriate model for describing the energy transfer in this system. The average distance between Dn and Ac moieties calculated from the conformational analysis (12 Å) is in very good agreement with the value estimated from the Förster equation (∼11 Å). At the same time, the calculated rate constant for energy transfer, averaged over multiple conformations of the system (3.6 ps), is in reasonable agreement with the experimental value (1.6 ps) estimated by transient absorption spectroscopy. The agreement between experimental results and computational data leads us to conclude that the energy transfer in Bi is well described by the Förster mechanism.
- This article is part of the themed collection: PCCP’s 15th anniversary
 Please wait while we load your content...
Please wait while we load your content...

