
Cobalt analogues of Roussin's red salt esters: a density functional study†
Abstract
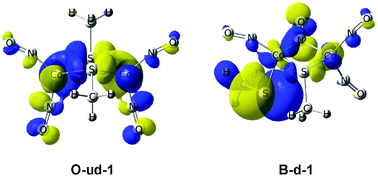
Density functional theory calculations on the Co2(NO)4(SR)2 compounds (R = CH3, CF3 and C4H9) predict butterfly and open isomeric structures with and without a direct Co–Co bond. The open Co2(NO)4(SR)2 structures are favored over the butterfly isomers, in terms of relative energy. Furthermore the open structures are predicted to have approximately twice as large HOMO–LUMO gaps than the butterfly Co2(NO)4(SR)2 isomers. For the related Co2(CO)6(SR)2 species, competing open and butterfly structures with similar HOMO–LUMO gaps were predicted. This could explain why the Co2(NO)4(μ-SR)2 compounds have already been synthesized and why no genuine Co2(CO)6(SR)2 derivatives have yet been reported.
 Please wait while we load your content...
Please wait while we load your content...

