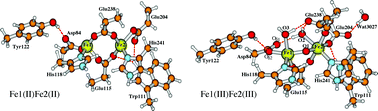
The R2 subunit of class-Ia ribonucleotide reductase (RNR) from Escherichia coli (E. coli) contains a diiron active site. Starting from the apo-protein and Fe(II) in solution at low Fe(II)/apoR2 ratios, mononuclear Fe(II) binding is observed indicating possible different Fe(II) binding affinities for the two alternative sites. Further, based on their Mössbauer spectroscopy and two-iron-isotope reaction experiments, Bollinger et al. (J. Am. Chem. Soc., 1997, 119, 5976–5977) proposed that the site Fe1, which bonds to Asp84, should be associated with the higher observed 57Fe Mössbauer quadrupole splitting (2.41 mm s−1) and lower isomer shift (0.45 mm s−1) in the Fe(III)Fe(III) state, site Fe2, which is further from Tyr122, should have a greater affinity for Fe(II) binding than site Fe1, and Fe(IV) in the intermediate X state should reside at site Fe2. In this paper, using density functional theory (DFT) incorporated with the conductor-like screening (COSMO) solvation model and with the finite-difference Poisson–Boltzmann self-consistent reaction field (PB-SCRF) methodologies, we have demonstrated that the observed large quadrupole splitting for the diferric state R2 does come from site Fe1(III) and it is mainly caused by the binding position of the carboxylate group of the Asp84 sidechain. Further, a series of active site clusters with mononuclear Fe(II) binding at either site Fe1 or Fe2 have been studied, which show that with a single dielectric medium outside the active site quantum region, there is no energetic preference for Fe(II) binding at one site over another. However, when including the explicit extended protein environment in the PB-SCRF model, the reaction field favors the Fe(II) binding at site Fe2 rather than at site Fe1 by ∼9 kcal mol−1. Therefore our calculations support the proposal of the previous Mössbauer spectroscopy and two-iron-isotope reaction experiments by Bollinger et al.

 Please wait while we load your content...
Please wait while we load your content...

