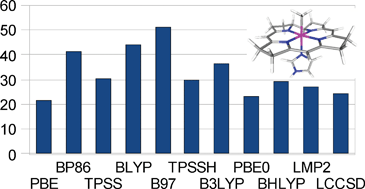
We have studied the homolytic dissociation of a methyl radical from a model of methyl cobalamin. For this reaction, density functional theory with an atom-pairwise dispersion correction (DFT-D) gives a dispersion contribution to the bond dissociation energy (BDE) of 22–51 kJ mol−1 depending on the functional, i.e. much more than common estimates for the total dispersion interaction energy of the methyl group in typical solvents. We show that this large energy correction results from many rather small (0–2 kJ mol−1) interactions that arise between the ligand and the metal and the other ligands when a short metal–ligand bond is formed. The energy terms result mostly from atom pairs connected by two or three bonds, i.e. terms that normally are ignored or scaled down at the molecular mechanics level, and have large contributions from r−8 terms. The added dispersion energy diminishes the variation in the calculated BDE observed among various generalised-gradient approximation (GGA) functionals, whereas a gap still persists between the results of GGA and hybrid functionals. Model calculations at the local MP2 and CCSD (second-order perturbation theory and coupled cluster theory with single and double excitations) levels are in a similar range as the dispersion interactions estimated by DFT-D (23–29 kJ mol−1). However, both the DFT-D and the wavefunction-based results include middle-range correlation effects that vary greatly between different DFT methods owing to their different density-based description in the short-range regime. Therefore, it is not meaningful to discuss which DFT method gives the most accurate estimate of the dispersion contribution to the BDE. Moreover, for a balanced treatment of dispersion during the binding reaction in solution, the dispersion energy of the ligand and the unbound complex with the surroundings needs also to be considered, which decreases the net dispersion contribution to binding by ∼20 kJ mol−1.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


