
Thermodynamic and kinetic properties of hydrogen defect pairs in SrTiO3 from density functional theory†
Abstract
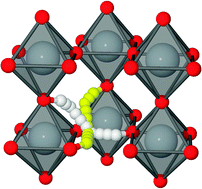
A density functional theory investigation of the thermodynamic and kinetic properties of hydrogen–hydrogen defect interactions in the cubic SrTiO3 perovskite is presented. We find a net attraction between two hydrogen atoms with an optimal separation of ∼2.3 Å. The energy gain is ca. 0.33 eV compared to two non-interacting H defects. The main cause of the net attractive potential is elastic defect interactions through lattice deformation. Two possible diffusion paths for the hydrogen defect pair are investigated and are both determined to be faster than the corresponding diffusion path for single hydrogen atoms. Finally, we set up a simple model to determine the contribution from the double hydrogen defect to the total hydrogen flux, and find the double defect to be the main diffusing species at temperatures below ca. 400 °C. Post submission infrared absorption experiments show excellent agreement with the proposed properties of the double hydrogen defect.
 Please wait while we load your content...
Please wait while we load your content...

