
A DFT based investigation into the electronic structure and properties of hydride rich rhodium clusters†
Abstract
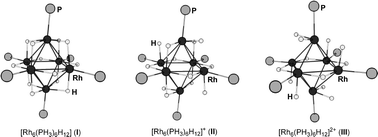
Density functional theory has been used to investigate the structures, bonding and properties of a family of hydride rich late transition metal clusters of the type [Rh6(PH3)6H12]x (x = 0, +1, +2, +3 or +4), [Rh6(PH3)6H16]x (x = +1 or +2) and [Rh6(PH3)6H14]x (x = 0, +1 or +2). The positions of the hydrogen atoms around the pseudo-octahedral Rh6 core in the optimized structures of [Rh6(PH3)6H12]x (x = 0, +1, +2, +3 or +4) varied depending on the overall charge on the cluster. The number of semi-bridging hydrides increased (semi-bridging hydrides have two different Rh–H bond distances) as the charge on the cluster increased and simultaneously the number of perfectly bridging hydrides (equidistant between two Rh centers) decreased. This distortion maximized the bonding between the hydrides and the metal centers and resulted in the stabilization of orbitals related to the 2T2g set in a perfectly octahedral cluster. In contrast, the optimized structures of the 16-hydride clusters [Rh6(PH3)6H12]x (x = +1 or +2) were similar and both clusters contained an interstitial hydride, along with one terminal hydride, ten bridging hydrides and two coordinated H2 molecules which were bound to two rhodium centers in an η2:η1-fashion. All the hydrides were on the outside of the Rh6 core in the lowest energy structures of the 14-hydride clusters [Rh6(PH3)6H14] and [Rh6(PH3)6H14]+, which both contained eleven bridging hydrides, one terminal hydride and one coordinated H2 molecule. Unfortunately, the precise structure of [Rh6(PH3)6H14]2+ could not be determined as structures both with and without an interstitial hydride were of similar energy. The reaction energetics for the uptake and release of two molecule of H2 by a cycle consisting of [Rh6(PH3)6H12]2+, [Rh6(PH3)6H16]2+, [Rh6(PH3)6H14]+, [Rh6(PH3)6H12]+ and [Rh6(PH3)6H14]2+ were modelled, and, in general, good agreement was observed between experimental and theoretical results. The electronic reasons for selected steps in the cycle were investigated. The 12-hydride cluster [Rh6(PH3)6H12]2+ readily picks up two molecules of H2 to form [Rh6(PH3)6H16]2+ because it has a small HOMO–LUMO gap (0.50 eV) and a degenerate pair of LUMO orbitals available for the uptake of four electrons (which are provided by two molecules of H2). The reverse process, the spontaneous release of a molecule of H2 from [Rh6(PH3)6H16]+ to form [Rh6(PH3)6H14]+ occurs because the energy gap between the anti-bonding SOMO and the next highest energy occupied orbital in [Rh6(PH3)6H16]+ is 0.9 eV, whereas in [Rh6(PH3)6H14]+ the energy gap between the anti-bonding SOMO and the next highest energy occupied orbital is only 0.3 eV. At this stage the factors driving the conversion of [Rh6(PH3)6H14]+ to [Rh6(PH3)6H12]2+ are still unclear.
 Please wait while we load your content...
Please wait while we load your content...

