
Water and hydroxide ion pathways in the σ-complexation of superelectrophilic 2-aryl-4,6-dinitrobenzotriazole 1-oxides in aqueous solution. A kinetic and thermodynamic study
Abstract
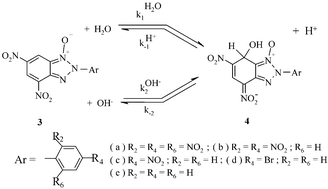
As part of our continuing studies of the highly electron-deficient nature of nitrobenzofuroxans, nitrobenzofurazans and related heterocycles, we report here a kinetic and thermodynamic study of σ-complexation for a series of 2-aryl-4,6 -dinitrobenzotriazole 1-oxides (3a–e) over a large pH range in aqueous solution. The reaction series represents a modulation in electrophilic properties of the benzotriazole moiety in formation of the corresponding hydroxy σ-adducts (4a–e). Analysis of the data has allowed dissection of observed rates into forward (kH2O1, kOH−2) and reverse (kH+−1, k−2) rate constants as well as the obtention of pKa values for H2O addition to the benzotriazole moiety. Our results reveal that 3a–e are superelectrophilic compounds with respect to 1,3,5-trinitrobenzene (TNB) as a standard electron-deficient aromatic, but less superelectrophilic compared to 4,6-dinitrobenzofuroxan (DNBF). Some data pertaining to buffer catalysis of the formation and decomposition of the adducts together with solvent deuterium isotope effects for these pathways are also reported. From these results, it is concluded that adduct formation occurs via general base catalyzed water attack: the general bases include notably H2O, HCO3−, CO32− as well as OH−. This contrasts with the situation for the σ-complexation of DNBF where HCO3− and CO32− were found to act as nucleophilic catalysts whereas OH− functioned as a general base catalyst. This contrasting behaviour provides further evidence that the dinitro-activated carbocyclic ring of the benzotriazoles 3a–e ranks somewhat lower in electrophilic/superelectrophilic properties compared to that in DNBF. Altogether, the results provide a basis for understanding the relationship between the superelectrophilic reactivities, as evidenced by the contrasting kinetic and thermodynamic properties of the systems at hand, and the varied abilities of these substrates to react in pericyclic Diels–Alder reactions.
 Please wait while we load your content...
Please wait while we load your content...