
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanical properties of cathode–electrolyte interphase layers in high-voltage lithium-ion batteries
Xiaotang
Shi
 a,
Andrew J.
Naylor
b,
Till
Fuchs
c,
Steffen
Schröder
c,
Franjo
Weber
a,
Anja
Henss
c,
Hans-Jürgen
Butt
a and
Rüdiger
Berger
*a
a,
Andrew J.
Naylor
b,
Till
Fuchs
c,
Steffen
Schröder
c,
Franjo
Weber
a,
Anja
Henss
c,
Hans-Jürgen
Butt
a and
Rüdiger
Berger
*a
aMax Planck Institute for Polymer Research, Mainz, 55128, Germany. E-mail: berger@mpip-mainz.mpg.de
bDepartment of Chemistry-Ångström Laboratory, Uppsala University, 751 21, Uppsala, Sweden
cInstitute of Experimental Physics I and Center for Materials Research, Justus Liebig-University Giessen, Heinrich-Buff-Ring 16, 35392 Giessen, Germany
First published on 17th April 2026
Abstract
LiNi0.5Mn1.5O4 (LNMO) is a promising cathode material owing to its high operating potential of 4.75 V vs. Li+/Li. However, the high potential triggers interfacial instability, such as electrolyte oxidation. A mechanically robust cathode–electrolyte interphase (CEI) is essential for maintaining structural integrity and ensuring reliable performance of high-voltage lithium-ion batteries. CEI layer thickness is often under 20 nm, making the assessment of its elastic properties challenging. We applied an amplitude-modulated/frequency-modulated scanning force microscopy method to enable quantitative mechanical characterization of thin CEI layers on rough composite electrode surfaces. We systematically varied the number of battery cycles and investigated the morphology and elastic modulus of the interphase layers on LNMO cathodes. The pristine crystalline LNMO surface exhibited an elastic modulus of approximately 126 ± 20 GPa, whereas the binder/carbon (b/c) regions had a modulus of 1.9 ± 0.1 GPa. After only 5 cycles the elastic modulus on LNMO decreased to 3.2 ± 1.2 GPa, indicating an LNMO passivation by CEI growth. After 200 cycles, the elastic modulus became homogeneous with the moduli on the LNMO and b/c regions reaching 3.9 ± 0.8 GPa and 3.9 ± 0.4 GPa, respectively. This mechanical convergence is supported by a convergence in chemical composition of the interphase between the LNMO and b/c regions. We also observed a compositional shift from ether-rich oligomers to a more oxidized, carbonyl-rich organic network. The final stabilized modulus of ≈4 GPa reflects an organic-dominated interphase with enhanced intermolecular interactions. Inorganic species are likely buried beneath the organic-rich top layer. This work provides understanding of interfacial stability and establishes a robust and reproducible framework for quantifying the elastic modulus of interphase layers within composite electrodes, providing insights for the design of stable high-voltage battery systems.
Introduction
The electrode–electrolyte interphase is critical for the performance of lithium-ion batteries.1 The interphase layer should remain intact on the surface of the active material to ensure stable cycling performance of batteries.2 During repetitive charge–discharge processes, the active materials undergo volume fluctuations, leading to mechanical stress and strain.3 These cyclic deformations, along with non-uniform strain distributions, often result in interphase cracking.4,5 Then the mechanical degradation of the interphase exposes fresh electrode surfaces to the electrolyte, accelerating decomposition reactions and promoting the continuous formation of interphase layers.6 This process ultimately increases cell impedance and reduces cycling performance.7,8 To prevent interphase cracking and mechanical degradation of the interphase, the mechanical properties of the interphase layer play an important role in maintaining structural integrity and mitigating brittleness during repeated volumetric fluctuations. This characteristic is essential for the interphase to remain mechanically elastic, enabling it to withstand the repeated stress generated during lithium ion de/intercalation processes. However, the thickness of an interphase layer is typically <20 nm. A mechanical characterization of the interface layer requires its deformation. For such thin interphase layer thicknesses, substrate deformation during indentation measurements becomes more likely, thereby compromising measurement accuracy. Therefore, the mechanical properties of thin interphase layers are difficult to quantify using traditional methods. We investigate the mechanical properties of the interphase layer formed on LiNi0.5Mn1.5O4 (LNMO) cathodes using the bimodal amplitude modulation-frequency modulation (AM-FM) mode in scanning force microscopy (SFM). This method enables the measurement of the elastic modulus of thin films due to small indentation depths <2 nm. These indentation depths enable quantitative, reliable results which are unaffected by the underlaying substrates.On the cathode side, the interphase is known as the cathode–electrolyte interphase (CEI). In contrast to the solid–electrolyte interphase (SEI) on anodes, CEI layers have been less studied. This is because the cathode surface remains relatively stable below 4.2 V in conventional carbonate-based electrolytes. As a result, significant interphase formation does not occur, making CEI improvements in these batteries less substantial.1 However, the pursuit of high-voltage cathodes to enhance energy density is often accompanied by reduced CEI stability.9 At elevated voltages, the electrolyte exceeds its thermodynamic stability limit, resulting in CEI formation with thicknesses ranging from a few nanometers up to approximately 20 nm.1 A uniform CEI layer functions as a protective barrier that suppresses parasitic reactions at high voltage. Most cathode materials are ceramic and brittle, making them particularly susceptible to stress and strain during high-voltage operation. This mechanical stress increases the likelihood of CEI cracking.10 When the CEI cracks or fails, structural and chemical degradation mechanisms can occur (Fig. 1a): (i) unprotected surfaces are exposed to the electrolyte thereby creating localized hot spots for electrolyte decomposition; (ii) products of electrolyte decomposition, such as hydrofluoric (HF) acid, can then directly attack the freshly exposed cathode surface and then promotes transition metal ions (Mx+) dissolution, the migrated Mx+ destabilizes the SEI, which in turn accelerates further electrolyte decomposition on the anode;11–13 (iii) ongoing interfacial reactions at the cathode drive inactive phases conversion, e.g., the conversion of the LNMO spinel to a poorly Li+ diffusion rock-salt-like phase.14 Reactions (i) and (ii) result in increased impedance and reactions (ii) and (iii) lead to capacity loss. An elastic CEI can better accommodate electrode volume changes, suppress crack propagation, and maintain intimate contact with active materials. These characteristics collectively contribute to a reduction of continuous electrolyte decomposition and the mitigation of capacity loss.15 Therefore, understanding the mechanical properties of the CEI is essential for evaluating interphase stability and improving the long-term performance of lithium-ion batteries.
 | ||
| Fig. 1 (a). The schematic diagram represents the degradation of cycling performance caused by cracking of the CEI layer and simultaneous deterioration of the SEI. (b) Schematic of the pristine cathode before cycling. (c) Electrode and formed interphase layer after the battery was cycled for multiple times. During cycling, electrolyte decomposition occurs near the electrode surface, leading to the formation of CEI layers on the crystalline LNMO surface. On the b/c regions, decomposition products also accumulate, forming interphase layers. We refer to the interphase formed on b/c areas as the non-CEI layer. (d) SFM measurements were performed on the electrode surface to characterize its morphology and mechanical properties. Prior to imaging, residual electrolyte was removed from the electrodes. | ||
One of the most important mechanical properties is Young's modulus, which defines how much strain a material experiences under a given stress.16 Numerous studies have quantified the Young's modulus of electrode materials using nanoindentation with a diamond indenter.17,18 For this purpose, a Berkovich geometry is widely used. It possesses a spherical cap with a radius exceeding 100 nm before transitioning into its pyramidal geometry.19 The modulus is typically extracted by fitting force–displacement curves. However, two factors have limited the application of nanoindentation for measuring the CEI layer.
The first factor is lateral resolution limitation which typically ranges from several hundred nanometers to a few micrometers.20 This resolution is insufficient for accurately mapping the nanoscale morphology of CEI layers. The limited resolution also hinders the differentiation between active materials and other components, such as binders and conductive carbon in composite electrodes (Fig. 1b). Daboss et al. observed that electrochemical strain predominantly occurred in the softer binder matrix rather than in the hard carbon microparticles. The authors concluded that the different ion dynamics would then affect later interphase formation.21 To differentiate the interphase layer on the cathode from that on the binder/carbon (b/c) surface, we refer to the interphase in the b/c region as the non-CEI layer (Fig. 1c).
The second factor is sensitivity due to a limitation in indentation depth. Reliable modulus measurements via nanoindentation generally require indentation depths of at least ≈15 nm.22 However, since the CEI typically ranges from a few to ≈20 nm in thickness, indentation depths of ≈15 nm risk substrate effects, thereby compromising measurement accuracy.16 Therefore, SFM methods which enable small indentation depths <2 nm are advantageous.
SFM is highly suitable for acquiring nanoscale information with minimal surface intrusion, typically limited to only a few nanometers.23 By correlating topography and modulus images, we differentiate the interphase layer formed on LNMO particles from that on the b/c region. Among SFM-based methods for mapping mechanical properties, tip-based nanoindentation24–26 and bimodal SFM27 are the two most widely used techniques. In tip-based nanoindentation, the elastic modulus is obtained by fitting force–displacement curves. Several studies have applied this method to SEI layers using indentation depths ranging from ≈5 nm to over 20 nm.28–31 Given that CEI layers are generally thinner than SEI layers, indentation depths must be limited to less than 2 nm for accurate indentation characterization (e.g., in a 20 nm-thick CEI).32 However, such shallow indentations are highly susceptible to inaccuracies arising from factors such as tip tilt, instrumental noise, and tip geometry.33,34
As an alternative, bimodal SFM enables rapid, high-resolution mapping of elastic properties by simultaneously exciting the cantilever at two resonance frequencies. The amplitude modulation-frequency modulation (AM-FM) technique combines the robust control of amplitude modulation at the first resonance with the high sensitivity and signal-to-noise ratio of FM at the second. AM-FM modulus mapping has been successfully applied to materials ranging from soft polymers (0.1–3 GPa) to hard metals (>100 GPa).35 AM-FM imaging typically offers higher speed compared to tip-based nanoindentation, as the elastic modulus is predominantly calculated by using the frequency shift of the second vibration eigenmode.36 In contrast, nanoindentation requires acquiring and fitting force–displacement curves at each point.37 The AM-FM technique is also suitable for thin samples because it quantifies small deformations with high sensitivity, with typical indentation depths ranging from 0.1 to 10 nm.35 This capability makes it particularly appropriate for the modulus mapping of CEI layers. To accurately map the modulus, one must account for surface tilt, as otherwise the indentation area may be misestimated.38 Height data obtained from the topography channel allows for pixel-by-pixel determination of local tilt angles, enabling correction of modulus values accordingly.
LiNi0.5Mn1.5O4 (LNMO) is a promising cathode material owing to its high operating potential (≈4.75 V vs. Li+/Li). However, the high potential triggers severe electrolyte oxidation and transition metal dissolution.39 Understanding the mechanical properties of CEI formed on LNMO is therefore essential to enhance interfacial stability and extend the cycle life of high-voltage lithium-ion batteries. Herein, we apply the AM-FM method to study CEI layers formed in high-voltage LNMO/Li cells. We track the evolution of CEI morphology and modulus in dependence of cycling number by recording AM-FM imaging on LNMO electrodes at different cycling stages (Fig. 1b–d). The modulus on LNMO particle surfaces decreases from ≈120 GPa to 3.2 GPa due to CEI formation. An increase in modulus observed in non-CEI regions suggests that interphase formation and mechanical evolution also take place on non-active components. Specifically, after 200 cycles, the moduli of the LNMO and b/c regions converge to ≈4.0 GPa. This mechanical convergence is supported by composition characterization, which reveals that the non-CEI progressively evolves toward a CEI-like chemical composition. Furthermore, compositional analysis reveals the chemical evolution of the interphase, and the relationship between the measured modulus and the interphase composition is also elaborated.
Results and discussion
LNMO composite electrode before cycling
In our work we use a composite electrode consisting of LNMO primary particles, polyvinylidene fluoride (PVDF) binder, and conductive carbon (Fig. 1b). The b/c regions exhibit a granular, aggregated structure, whereas the LNMO particles display distinct crystalline surfaces with a diameter of several micrometers. In the pristine state, the electrode surface has well-defined LNMO crystals alongside localized binder-carbon aggregates. We first analyze the pristine LNMO electrode before examining CEI formation after cycling (Fig. 2a–c). The topography reveals two main types of surface features: well-defined crystalline particles (black-bordered area) and b/c aggregates (white-boarded area) (Fig. 2a). Owing to the different orientation of LNMO crystals, angle calibration is essential to correct the measured modulus for obtaining accurate mechanical properties of interphase layers. We calibrated the AM-FM method by using silica microspheres. A correction factor was obtained for different indentation angles enabling accurate calibration of modulus values across the electrode surface. Details of the calibration procedure are given in the Experimental section. In addition, several small particles were observed on the crystalline surface. The small particles in region I (Fig. 2b) exhibit a significantly lower modulus than the surrounding crystalline surface, suggesting that they are b/c aggregates. Apart from these observations, the crystalline surfaces appear clean. To verify this assignment, scanning electron microscopy-energy-dispersive X-ray spectroscopy (SEM-EDS) mapping was performed on the pristine electrode surface (Fig. S1). The elemental distributions show that Ni, Mn, and O are localized on the crystalline particles, whereas C and enriched F from the PVDF binder are predominantly distributed in the interstitial particle aggregates regions, thereby providing chemical validation for distinguishing LNMO particles from b/c regions in the SFM analysis. For the crystalline surfaces, we manually segmented regions labeled I through III based on the topographic outline of crystal facets and plotted the corresponding modulus distributions (Fig. 2c). We use the standard deviation sigma (σ) obtained from the Gaussian fit to represent the distribution width and the fitting error. The moduli at regions I, II, and III correspond to 145 ± 10, 113 ± 12, and 117 ± 11 GPa, respectively. These values are consistent with previously reported modulus values for LNMO (136 ± 12 GPa) in the literature.40 The variation in modulus between regions I and II, III may result from anisotropic atomic arrangement and bonding associated with different crystallographic orientations.41,42 The LNMO-free electrode reveals clear aggregates of binder and carbon (Fig. S2a). The modulus distribution for b/c mixture has a peak value of 1.9 ± 1.0 GPa (inset of Fig. 2c). The modulus histogram of b/c mixture shows a long tail, which arises because the conductive carbon phase exhibits a higher modulus than the polymer binder. As carbon particles are embed into the binder matrix, local variations in binder thickness contribute to a broader modulus distribution, particularly above 4 GPa. Such prepared LNMO composite electrodes were used in coin cells, and then cycled for varying times to promote interphase layer formation. Therefore, the pristine LNMO electrode and b/c electrode serve as a reference for identifying and distinguishing the CEI layer and non-CEI regions during interphase formation and evolution at different cycling stages. | ||
| Fig. 2 Modulus evolution of interphase layers observed with SFM using amplitude modulation-frequency modulation. SFM characterization of the surface of a pristine LNMO electrode (a–c) and electrode after 5 (d–f), 30 (g–i), 60 (j–l) and 200 (m–o) cycles: (a, d, g, j and m) topography images, (b, e, h, k and n) calibrated modulus maps, and (c, f, i, l and o) corresponding modulus distribution histogram for the masked regions (I–IV and b/c). The black dotted curve represents Gaussian fits of each modulus. The histograms of the entire unmasked maps correspond to the gray-bordered bar chart. Scale bar: 2 µm. | ||
Interphase layer on LNMO electrode after cycling
We cycled LNMO/Li batteries for certain times to obtain interphase layers at different stages. After 200 cycles, the cell retained approximately 81% of its initial capacity (Fig. S3a), which is comparable to values reported in the literature for similar electrolyte systems.39 The first discharge capacity was 130.3 mAh g−1 and increased to 132.6 mAh g−1 after two cycles. This initial capacity increase can be attributed to an activation process during early cycling. During the initial cycling stage, electrolyte wetting and pore penetration are incomplete. The interphase layer is still forming due to electrolyte decomposition. As these transient processes stabilize after the first few cycles, the discharge capacity exhibits a slight rebound, reflecting improved electrolyte accessibility and relative stabilization of the electrode–electrolyte interface.43 The voltage gap between redox plateaus (Fig. S3b) rises with increasing current density. At a constant C-rate of 0.5C, the voltage gap between redox plateaus in the capacity vs. voltage curves gradually increases from the 30th to the 200th cycle. This progressive voltage hysteresis indicates increasing polarization during cycling, which is commonly associated with slower ion transport, hindered charge-transfer kinetics, structural degradation of the electrode, and the accumulation of interfacial side reactions.44After charge–discharge cycling LNMO electrodes in coin cells, the AM-FM method was employed to map the surface morphology and the modulus of the top layer (Fig. 2d, e, g, h, j, k, m and n). The moduli were analyzed in corresponding histograms (Fig. 2f, i, l and o). After five cycles, the primary LNMO surfaces remained clearly visible, showing distinct crystalline facets and sharp grain boundaries (Fig. 2d). The modulus of the LNMO surface decreased significantly from over 100 GPa to several GPa owing to the formation of the CEI layer (Fig. 2e). The high-voltage cycling induces substantial CEI growth and pronounced chemical evolution, resulting in interphase formation.45 In contrast, any effects arising from short-term electrolyte exposure are limited as they may induce only weak surface reactions or solvent adsorption. The adsorbed electrolyte solvents do not undergo significant oxidative decomposition. So, mere exposure of cathode materials to the electrolyte only leads to weak and nascent interphase layers.45,46 Thus, the interphase layer formed solely through passive electrolyte exposure contributes negligibly to the overall interphase formation, and therefore we will not discuss this effect here. The modulus map reveals differences between the LNMO particles and b/c regions. In regions containing LNMO particles, which are labeled by I–IV, the CEI layer exhibited a higher modulus than the surrounding b/c area. The modulus values in regions I–IV ranged from 2.3 ± 0.7 to 3.5 ± 0.9 GPa (Fig. 2f), suggesting that CEI stiffness varies with crystallographic orientation and surface termination.47 Furthermore, the width of the histogram indicates differences in modulus values owing to local variations in CEI composition and thickness.48 The modulus variation across region II suggests the formation of a mechanically heterogeneous CEI layer. An enlarged view (Fig. S4a–c) highlights the specific area selected for detailed analysis. The topography image (Fig. S4b) reveals that the early-stage interphase structure exhibits nanometric secondary particles distributed across the crystalline surface. These secondary particles indicate that the electrolyte decomposition products have coated the underlying crystalline surface with a non-uniform thickness and increased surface roughness. The corresponding phase image (Fig. S4c) further demonstrates mechanical heterogeneity, which is consistent with compositional and structural variations within the CEI layer during the early stage of formation. In the b/c region, the calibrated modulus slightly decreased from 1.9 ± 1.0 to 1.4 ± 1.2 GPa due to the formation of a non-CEI surface layer. Li et al. reported that the b/c mixture reacts with the electrolyte, leading to the formation of a soft, organic-rich surface film containing fluorinated organics and carbonate fragments.49 Daboss et al. observed that local deformation predominantly occurs in the binder matrix rather than in the active hard carbon particles, indicating distinct electrochemical behavior between the binder and the active material.21 Such differences in ion accessibility and electrochemical strain may promote the formation of compositionally distinct interphase layers on the binder and on the active material after cycling. In conclusion, cycling the LNMO electrode 5 times already resulted in a significant decrease in moduli compared to the pristine surface.
We continued to measure the surface topography and modulus of the LNMO electrode after 30 cycles (Fig. 2g and h). With this extended cycling, the CEI composition evolves, as the amount of organic components from electrolyte oxidation in the CEI increase and transition-metal dissolution products (e.g., MnFx) incorporate into the interphase.9,50 The modulus distribution of manually segmented regions shows 2.7 ± 0.1, 6.5 ± 0.7, 2.0 ± 0.1 and 5.1 ± 0.4 GPa in the CEI layer (region I–IV) and 1.8 ± 0.8 GPa in the non-CEI layer (Fig. 2i). The non-CEI layer still displayed a lower modulus than the CEI layer formed on the surface of LNMO crystalline (Fig. 2h). The slight increase of the mean value from 1.4 ± 1.2 to 1.8 ± 0.8 GPa may result from the migration of transition metal compounds from the LNMO surface to the carbon/binder region during continued cycling.49 In comparison to the electrode after 5 cycles (σ of 0.4 to 1.2 GPa), the modulus distribution in region I and III (σ of 0.1 GPa) narrowed across crystal planes, which suggests more uniform mechanical properties of the local CEI layer. This uniformity may result from the merging of CEI layers across neighboring particles or from the significant accumulation of electrolyte decomposition products, both of which contribute to a mechanically homogenized CEI. These observations are consistent with the progressive growth and evolution of the CEI during cycling.51 Regions I and III exhibit lower modulus values (2.7 and 2.0 GPa) compared to regions II and IV (6.5 and 5.1 GPa), indicating a heterogeneous CEI structure composed of both rigid and soft components. This difference may originate from partial CEI dissolution into the electrolyte and deposition of transition-metal dissolution products into the interphase.
As cycling progresses, the interphase layer typically becomes thicker due to the exposure of fresh cathode surfaces and continued electrolyte decomposition.52 Therefore, additional SFM measurements were conducted on the LNMO electrode after 60 cycles (Fig. 2j and k). Compared to electrodes cycled for 5 and 30 cycles, the elastic modulus map (Fig. 2k) shows minimal variation across different crystal planes. Mechanical properties become more uniform. The CEI layer at this stage appears amorphous with a morphology characterized by rounded particles in region I (Fig. S5), suggesting the formation of a thicker and mechanically consistent interphase. The manually segmented regions exhibit modulus values of 4.2 ± 0.2, 4.9 ± 1.2, 5.3 ± 1.6 and 4.2 ± 0.3 GPa in regions I–IV (Fig. 2l). The inserted image (Fig. 2l) shows the modulus distribution of the non-CEI region. The modulus of the non-CEI layer increased from 1.8 ± 0.8 GPa to 4.7 ± 1.3 GPa after extended cycling, converging to the modulus of the CEI layer. This result suggests that the b/c region becomes fully covered with electrolyte decomposition products, similar to the coverage on the LNMO surface.
To investigate the long-term evolution of the interphase modulus, we recorded the surface topography (Fig. 2m) and the calibrated modulus map (Fig. 2n) of the LNMO electrode after 200 cycles, and plotted the modulus histogram for each region (Fig. 2o). The mean value of the modulus of the non-CEI layer corresponded to 4.2 ± 1.1 GPa and remained close to the modulus of the adjacent CEI layer. This observation confirmed that an interphase layer with similar composition to CEI had also formed on the surface of the binder and carbon. Additionally, the modulus histogram displays a tail in the high-modulus region, which may result from the incorporation of transition-metal ion products originating from the LNMO surface. The segmented regions exhibit modulus values of 3.6 ± 0.1, 3.7 ± 0.2, 4.6 ± 0.5 and 4.6 ± 0.5 GPa in regions I–IV (Fig. 2o). Consistent with the contrast observed after 60 cycles, the modulus distribution across regions became narrower compared to that recorded after 5 and 30 cycles. This small variation indicates that CEI layers maintain mechanical uniformity even after extended cycling.
We then conducted conventional nanoindentation measurements for comparison to support the methodological validity of our results (Fig. S6). Due to the inherent constraints of the Berkovich indenter geometry and contact mechanics models, conventional measurements required a minimum indentation depth of 30–100 nm on cycled electrodes. These indentation depths significantly exceed the CEI layer thickness, which is approximately 20 nm. Consequently, the mechanical properties of the underlying metal oxide substrate interfere with the measurement, leading to an overestimated modulus (e.g. ≈10.3 GPa on LNMO). Furthermore, conventional nanoindentation has limited lateral resolution (≈1.25 µm) and fails to distinguish between LNMO particles and binder/carbon regions. By contrast, the AM-FM technique is well suited for thin-film systems because indentation depths can be reduced to below 2 nm, enabling reliable modulus mapping of CEI layers without significant substrate influence. Its high lateral resolution not only allows clear distinction between LNMO particles and b/c regions, but also provides topography information. This capability enables pixel-by-pixel determination of local surface tilt angles, allowing for the correction of modulus values (Fig. 2b, e, h, k and n). The intrinsic elastic modulus of the CEI was determined to be around 4 GPa after 200 cycles.
We summarize and discuss the elastic moduli of both the CEI and non-CEI layers at different cycle numbers, including moduli obtained from the pristine LNMO electrode (Fig. 3a). We calculated mean modulus values and the corresponding standard deviations (σ). The standard deviation arises from two primary contributions: measurement uncertainty and intrinsic mechanical inhomogeneity of the interphase layer within each region. The intrinsic mechanical inhomogeneity of the interphase originates from its heterogeneous composition, which consists of varying proportions of organic and inorganic components as well as local CEI thickness.48 Measurement-related contributions include frequency-tracking noise and thermal drift of the SFM system. In principle, the relative contribution of measurement error to σ can be evaluated by comparing σ values across different samples or across different regions within the same sample. In this study, the smallest σ observed is approximately 0.1 GPa. So, the σ arises mainly from interphase mechanical heterogeneity rather than instrumental noise under the optimized experimental conditions employed. We also calculated the mean modulus and standard deviation separately for crystalline LNMO regions and b/c regions for each sample (Table 1). This parameter reflects the dispersion of measured modulus values within each distinct surface region. The clean crystalline LNMO surface and b/c mixture exhibited modulus values of 126 ± 20 GPa and 1.9 ± 0.1 GPa, respectively. Average modulus values with corresponding standard deviations of the CEI layer after 5, 30, 60, and 200 cycles were 3.2 ± 1.1, 4.5 ± 1.6, 4.1 ± 0.7, and 3.9 ± 0.8 GPa, respectively. In the LNMO regions, this dispersion is primarily attributed to variations in crystal orientation.48 As for the b/c regions, they exhibit the lowest modulus dispersion prior to cycling. However, the dispersion in the b/c regions increases after cycling. We attribute this increase to microscale heterogeneity leading to local variations in ionic diffusivity. This dispersion promotes non-uniform interphase formation across the b/c network.53 The observed CEI modulus values are reasonable, as CEI layers which formed under cycling conditions are often much softer than pure inorganic layers8 (e.g., LiF and Li2CO3), due to their porosity and the incorporation of organic species. Organic and inorganic species, like oligomers and Li2CO3 are thermodynamically unstable at around 4 V vs. Li/Li+. Nevertheless, oligomeric species and Li2CO3 persist within the CEI under cycling conditions owing to kinetic limitations54 and a dynamic regeneration process.55 Both effects result in the retention of these species within the CEI rather than their complete oxidative removal during battery cycling. The initial increase in modulus from 3.2 to 4.5 GPa likely reflects interphase densification or an increase in inorganic content during early CEI formation. This interpretation is consistent with the findings of previous studies,49 who observed progressive accumulation of inorganic species from the first few formation cycles up to 20 cycles. An alternative explanation is that the initial organic-rich CEI is mechanically unstable and prone to cracking. This instability may lead to localized stiffening due to structural reorganization and the redistribution of inorganic species. The relatively high standard deviation of 1.6 GPa after 30 cycles further suggests modulus non-uniformity across different crystalline facets. Based on the above analysis, we conclude that the interphase exhibits pronounced heterogeneity (Fig. 3b) after the initial pre-cycled stages (5 and 30 cycles). These results indicate that the CEI behaves as a dynamic and non-uniform layer during the early stages of cycling. Initially, this lateral heterogeneity (Fig. S4c) indicates localized electrolyte decomposition, which leads to the formation of a chemically mixed and unstable CEI that is unevenly distributed across the crystalline surface. Partial CEI dissolution into the electrolyte and deposition of transition-metal species within the interphase further contribute to spatial variations in modulus. In situ SFM imaging (cycling up to 4.5 V) shows that CEI films form mainly at the edge planes of LiCoO2 crystals and decompose upon discharge, while little film growth occurs on the basal planes.56 Similarly, CEI formation on LNMO particles strongly depends on the specific crystallographic planes exposed at early stage.47
 | ||
| Fig. 3 (a) Calibrated moduli of pristine LNMO and interphase layers formed after 5, 30, 60 and 200 cycles. The error bars correspond to the standard deviation extracted from Gaussian fits. (b) Schematic illustration of the evolution of the modulus within the interphase layer on both the LNMO surface and the b/c region, including the modulus of the different components present in the pristine electrode. | ||
| Pristine | 5 cycles | 30 cycles | 60 cycles | 200 cycles | |
|---|---|---|---|---|---|
| LNMO (GPa) | 126 ± 20 | 3.2 ± 1.1 | 4.5 ± 1.6 | 4.1 ± 0.7 | 3.9 ± 0.8 |
| b/c (GPa) | 1.9 ± 0.1 | 1.6 ± 0.3 | 2.2 ± 0.7 | 3.5 ± 1.1 | 3.9 ± 0.4 |
After 60 cycles, both the average modulus and standard deviation decreased to 4.1 GPa and 0.7 GPa, respectively. After 200 cycles, the modulus stabilized at approximately 3.9 GPa (Table 1). This downward trend in the standard deviation indicates mechanical homogenization (Fig. 3b) upon extended cycling (60–200 cycles), likely resulting from the formation of a more elastic and continuous CEI layer, which evolves from a heterogeneous, compositionally diverse interphase in the early stages. Such evolution is consistent with progressive CEI degradation and reorganization driven by electrolyte oxidation. The improvement in uniformity results from ongoing electrolyte decomposition and transition-metal dissolution, which together produce a re-evolved and compositionally mixed CEI.57
For the non-CEI regions, the modulus initially decreases slightly from 1.9 ± 0.1 GPa to 1.6 ± 0.3 GPa after 5 formation cycles (Fig. 3a). This decrease is likely due to the deposition of organic electrolyte decomposition products. The subsequent increase to 2.2 ± 0.7 GPa suggests increased incorporation of inorganic components and densification of the non-CEI layer. After 60 and 200 cycles, the modulus of the non-CEI region stabilizes at 3.5 ± 1.1 GPa and 3.9 ± 0.4 GPa, respectively, showing a similar trend to that of the CEI layer. This convergence in mechanical behavior (Fig. 3b) indicates that the non-CEI region also undergoes stabilization, consistent with previous studies reporting that electrolyte decomposition products in inactive regions become mechanically stable during prolonged cycling.49 This mechanical convergence is further elucidated by time-of-flight secondary ion mass spectrometry (ToF-SIMS) depth profiling to track the chemical evolution of the interphase. The NiO2−- and C2−-fragments (Fig. S7) enable a reliable spatial distinction between LNMO particle regions and b/c regions. C2HO− fragments result from organic electrolyte decomposition and exhibit similar depth distributions in both LNMO and b/c regions across different cycling stages (Fig. S8a–c). PO3− fragments are formed as decomposition products from the conducting salt LiPF6. Across all samples the signal intensity of PO3− is higher in the LNMO region than in the b/c region; however, the difference decreases significantly from 0.18 to 0.09 after 200 cycles (Fig. S8d–f). This reduction directly correlates with the decreasing modulus gap between the interphase on LNMO and b/c regions, from 3.2 ± 1.1 GPa vs. 1.6 ± 0.3 GPa at early stages to 3.9 ± 0.8 GPa vs. 3.9 ± 0.4 GPa after long-term cycling. Furthermore, the gradual increase of NiO2− intensity in the b/c region (Fig. S8d–f) suggests a non-uniformly covering CEI, which allows for transition metal dissolution and redeposition under high-voltage conditions. After 200 cycles, the interphase layer on both LNMO and b/c regions develop a characteristic layered CEI-like structure, which shows an organic-rich outer layer and an inorganic-rich inner layer (green lines in Fig. S8). Overall, the ToF-SIMS results indicate that the interphase in the b/c regions progressively evolves toward a CEI-like composition, providing evidence for the observed mechanical convergence.
During the early cycling stage, the heterogeneous CEI remains unstable. Both the softer (≈2 GPa) and harder (≈6 GPa) regions progressively evolve toward an intermediate modulus of ≈4 GPa. The initially lower modulus can be enhanced through further oxidation of electrolyte products and the deposition of inorganic species, which improves mechanical stability but simultaneously increases interphase impedance due to the accumulation of inorganic compounds (such as MnFx and LiF). This process of mechanical stabilization generally emerges after approximately 60 cycles. Unlike the formation of a thin, continuous, and ordered interfacial protection layer of dissolved Cu and Fe ions in hydronium-ion batteries,58 the redeposition of dissolved transition-metal ions of Mn and Ni tends to occur on the electrode surface in a heterogeneous and poorly ordered manner. This disordered redeposition contributes to an interphase with higher ionic resistivity. Consequently, these deposits at the electrode surface can severely impede Li+ transport kinetics and accelerate electrolyte oxidation at the exposed active sites.
To directly study the accompanying evolution of ion-transport and charge-transfer kinetics associated with this mechanically stabilized interphase, we performed electrochemical impedance spectroscopy (EIS) measurements on LNMO/Li cells after 5, 30, 60, and 200 cycles (Fig. S9a). The Nyquist plots consist primarily of two semicircles, which we attribute to the solid electrolyte interphase film resistance (Rf) in the high-frequency region and the charge-transfer resistance (Rct) in the medium-frequency region. We used the equivalent circuit model (inset in Fig. S9a) to fit the impedance data. The impedance spectra reveal that Rf increased from 61 Ω to 86 Ω after 60 cycles, and further increased to 104 Ω after 200 cycles (Fig. S9b), where CEI modulus stabilized at around 4 GPa. This trend provides direct evidence for increased ionic transport resistance which is related to interphase layer evolution. Consequently, the extracted Rct increased with cycling and reached as high as 347 Ω after 200 cycles. This increased Rct restricted interfacial charge-transfer kinetics after long-time cycles. It is also noteworthy that both Rf and Rct at the 5th cycle are higher than those at the 30th cycle. The decrease in both values is consistent with the formation of a chemically unstable and continuously evolving interphase during the early cycling stage. A more mature SEI formed upon subsequent cycling which facilitates Li+ de-solvation and interfacial charge transfer, leading to reduced impedance.59 The EIS results complement our findings. As the mechanical homogeneity improved with cycle numbers, the interphase layer impedes Li+ transport properties and slows electrochemical kinetics. The latter leads to the degradation of capacity retention and rate capability during extended cycling.60
To establish a direct link between the observed mechanical changes and the underlying chemical origin of the CEI, we studied the compositional changes of the interphase at different cycling stages using X-ray photoelectron spectroscopy (XPS). XPS spectra were collected from the LNMO electrode before and after 5, 30, 60, 200 cycles (Fig. S10). In the C 1s spectra, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C signal originates primarily from the conductive carbon. The measurements exhibit a gradual decrease in CC relative intensity after cycling, which indicates a decomposition reaction of the electrolyte on the surface of conductive carbon, leading to interphase formation on its surface.61 After 60 cycles, the CC peak exhibits an increase in binding energy of approximately 0.3 eV, associated with the formation of an electric potential gradient between the cathode material and the CEI.62 This peak shift and the further decrease in intensity after 200 cycles indicate the formation of a distinct interphase layer covering the conductive carbon surface at this stage. The C–C/C–H, C–O, and CO signals result from products of the organic electrolyte decomposition. To quantitatively describe the C–C/C–H, C–O and CO evolution, we calculated the total peak area of these three components and the intensity ratio of CO to C–O bonds (Fig. 4a). The total organic peak area decreased slightly from 5 to 60 cycles and then increased substantially after 200 cycles, reflecting the combined effects of component decomposition, dissolution, and regeneration. Meanwhile, the CO to C–O ratio increased with cycling, indicating a transformation from initially formed ether-rich oligomers to a more oxidized carbonyl network. This interpretation is supported by the signals corresponding to O 1s: the ratio of CO/PO to C–O/P–O in O 1s spectra decreased slightly after 30 cycles and then increased after 60 and 200 cycles. Such a compositional shift is expected to increase the modulus within the organic CEI components.
C signal originates primarily from the conductive carbon. The measurements exhibit a gradual decrease in CC relative intensity after cycling, which indicates a decomposition reaction of the electrolyte on the surface of conductive carbon, leading to interphase formation on its surface.61 After 60 cycles, the CC peak exhibits an increase in binding energy of approximately 0.3 eV, associated with the formation of an electric potential gradient between the cathode material and the CEI.62 This peak shift and the further decrease in intensity after 200 cycles indicate the formation of a distinct interphase layer covering the conductive carbon surface at this stage. The C–C/C–H, C–O, and CO signals result from products of the organic electrolyte decomposition. To quantitatively describe the C–C/C–H, C–O and CO evolution, we calculated the total peak area of these three components and the intensity ratio of CO to C–O bonds (Fig. 4a). The total organic peak area decreased slightly from 5 to 60 cycles and then increased substantially after 200 cycles, reflecting the combined effects of component decomposition, dissolution, and regeneration. Meanwhile, the CO to C–O ratio increased with cycling, indicating a transformation from initially formed ether-rich oligomers to a more oxidized carbonyl network. This interpretation is supported by the signals corresponding to O 1s: the ratio of CO/PO to C–O/P–O in O 1s spectra decreased slightly after 30 cycles and then increased after 60 and 200 cycles. Such a compositional shift is expected to increase the modulus within the organic CEI components.
 | ||
| Fig. 4 Quantitative analysis of CEI components in dependence on cycling number. (a) The peak area evolution of organic electrolyte decomposition products, the ratio dependence of CO to C–O in C 1s spectra, and CO/PO to C–O/P–O in O 1s spectra. (b) Peak area dependence of LiF and P–F bonding in the F 1s spectra. | ||
However, the CEI layer is composed of both organic and inorganic components. Thus, the modulus of the interphase layer should not only depend on changes in the organic deposits. In the F 1s spectra, the P–F and LiF signals originate from inorganic lithium-salt decomposition products, which generally possess higher moduli than organic carbonate-derived species. For example, crystalline LiF exhibits a modulus as high as approximately 89 GPa.8 Accordingly, we quantified the evolution of LiF and P–O peak areas as a function of cycle number (Fig. 4b). After 30 cycles, the peak area of the LiF signal reached a maximum. LiF is chemically stable and resistant to further decomposition.63 With continued cycling, electrolyte solvent decomposition continued, and the peak area of the LiF signal decreased due to the increase in organic CEI components. However, we cannot exclude that the decrease in LiF content after long-time cycles may also result from LiF detachment as a result of CEI decomposition.45,64 In summary, we conclude that a LiF-richer layer might be present as an inner layer of the CEI after a higher cycling numbers. A LiF-richer inner layer would contribute less to the measured surface modulus.
Considering a continuously evolving CEI morphology and modulus values of only several gigapascals, the CEI can be identified as being dominated by organic compounds. In this structure, LiF does not form a continuous, high-modulus inorganic framework. So, there is a limited influence on the overall surface modulus. The interphase modulus evolved from a relatively low value associated with C–O rich compositions after the initial 5 cycles (Table 1). The increased ratio between CO/C–O leads to stronger molecular polarity, which suggests enhanced intermolecular interactions. The intermolecular interactions restrict segmental mobility, leading to an increased mechanical modulus within the organic interphase layer. A relatively higher modulus exists owing to less organic electrolyte decomposition and more LiF after 30 cycles. Finally, the stabilization at approximately 4 GPa arises from the combined effects of increased organic oxidation and the reduced contribution of LiF to the surface-sensitive modulus measurement.
To gain insights into the thickness of the CEI at different cycling stages, several complementary approaches were explored. SFM scratching experiments suggest a CEI thickness on the order of 15 nm, while cross-sectional transmission electron microscopy (TEM) observations indicate an apparent interphase thickness exceeding 20 nm. XPS spectra of cycled electrodes still exhibit detectable metal-oxide signals, implying that the CEI is porous rather than fully dense. However, these thickness estimates are subject to substantial uncertainties due to incomplete CEI removal during SFM scratching, projection and crystallographic orientation effects in TEM imaging, and the limited applicability of signal attenuation analysis for porous interphases. These approaches provide only order-of-magnitude estimates. As a result, these thickness estimates cannot be directly compared with surface-sensitive elastic modulus measurements quantitatively.
Conclusion
We measured the mechanical properties of the interphase layer formed on LiNi0.5Mn1.5O4 (LNMO) cathodes by using the bimodal amplitude modulation-frequency modulation (AM-FM) mode in scanning force microscopy (SFM). The lateral resolution enabled us to distinguish different components in composite electrodes and record the height information of each pixel. Owing to the different orientation of LNMO crystals, we calibrated the AM-FM method by using silica microspheres. A correction factor was obtained for different indentation angles enabling accurate calibration of modulus values across the electrode surface. By combining AM-FM technology with tilt-angle calibration, we are able to apply this approach to characterize various cathode materials and obtain accurate, quantitative modulus maps of their CEI layers.The measured evolution in modulus reflects an initial CEI passivation process, followed by subsequent reformation. A key finding is the progressive mechanical homogenization of both the CEI on the LNMO region, and non-CEI on binder/carbon (b/c) region, which eventually converged to a modulus of approximately 4 GPa during long-term cycling. ToF-SIMS depth profiling provides the chemical evidence for this mechanical convergence. The chemical contrast between LNMO and b/c regions significantly decreased as both areas evolved toward a similar layered interphase. This finding confirms that the interphase on b/c regions progressively evolves toward a CEI-like chemical composition. XPS analysis reveals that the modulus evolution was driven by a compositional shift from ether-rich oligomers to a more oxidized carbonyl network. The final stabilized modulus reflects an organic-dominated interphase. EIS measurements further correlate the mechanical homogenization with an increase in both interphase and charge-transfer resistances.
It is well established that CEI exhibits depth-dependent compositional heterogeneity.65 Therefore, the change in modulus is not determined by thickness alone. Instead, it is driven by compositional changes at different depths. AM-FM measurements primarily probe the outer CEI due to the shallow tip–sample interaction. The progressive removal of the outer CEI can be achieved by systematically increasing the tip–sample interaction force and adjusting scanning parameters. Subsequent AM-FM measurements on these modified surfaces increasingly reflect the mechanical properties of the inner CEI. Such depth-resolved investigations of the CEI modulus and structure may contribute to a deeper understanding of its functional mechanisms and provide useful guidance for the future design of the CEI in high-voltage cathode systems.
Experimental details
Preparation of the LNMO electrode. The LNMO slurry was prepared by mixing LiNi0.5Mn1.5O4 (LNMO) (RONBAY TECHNOLOGY), poly(vinylidene fluoride) (PVDF, >99.9%) (Xiamen Tmax), and conductive carbon (Xiamen AOT, battery grade) in N-methyl-2-pyrrolidone (NMP) (Carl Roth, >99.8%) at a mass ratio of 90
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5. The mass ratio of PVDF to NMP was 1:16. The slurry was stirred for 8 hours at 800 rpm, then cast onto aluminum foil, blade-coated, and dried at 80 °C for approximately 12 hours. The dried electrode was punched into circular disks with a diameter of 14 mm, resulting in an active material loading of approximately 4.2 mg cm−2.
:5:5. The mass ratio of PVDF to NMP was 1:16. The slurry was stirred for 8 hours at 800 rpm, then cast onto aluminum foil, blade-coated, and dried at 80 °C for approximately 12 hours. The dried electrode was punched into circular disks with a diameter of 14 mm, resulting in an active material loading of approximately 4.2 mg cm−2.
Assembly of coin cells. 2032-type coin cells were assembled in an argon-filled glovebox (MBRAUN; p(H2O)/p ≤ 0.1 ppm, p(O2)/p ≤ 0.1 ppm), using the LNMO cathode, polypropylene (PP) separator (PP-25, Celgard), lithium foil counter electrode, and 60 µL of electrolyte. The electrolyte (Sigma-Aldrich, battery grade) contained 1 mol LiPF6 in a 1
:1 (v/v) mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). Pure dimethyl carbonate (DMC, 99%, Sigma-Aldrich) was used to remove residual lithium salts. After assembly, the cells were removed from the glovebox and allowed to rest for 8 hours before electrochemical testing.
Polystyrene (PS) film sample. A PS solution (40 mg mL−1) was prepared by dissolving polystyrene (molar weight = 110
843 g mol−1) in tetrahydrofuran. Then, 60 µL of the PS solution was dropped onto a glass substrate. A uniform PS film was obtained by using a spin coater (LabSpin6, SUSS MicroTec) operated at 2000 rpm, with an acceleration of 400 rpm s−1 and a spin time of 30 seconds. The coated film was then left in a fume hood at room temperature for 3 hours. The final film thickness was approximately 500 nm.
Silica microspheres sample. A silica microsphere (SiO2) dispersion (SS04N, Bang Laboratories, Inc.; mean radius ≈1.5 µm) was diluted with ethanol. Then, the diluted dispersion was dropped onto the prepared PS film. The sample was kept at room temperature for 10 minutes, followed by drying in an oven at 110 °C for 20 minutes.
 | ||
| Fig. 5 Schematic illustration of the working principle of AM-FM mode. A1,free and A2,free represent the free amplitudes of the first and second resonance modes, respectively. A1 denotes the instantaneous amplitude of the first mode. Δf2 indicates the resonance frequency shift of the second mode. | ||
The theory of elastic modulus quantification used in AM-FM mode has been described previously.35,36 As the tip radius is much smaller than the contact radius, the SFM tip is modeled as a flat punch. Based on this assumption, the elastic modulus E was calculated using the following formula:

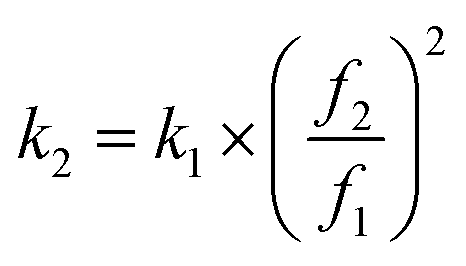 . Upon approaching the sample, the interaction phase Φ1 was determined at the first resonance frequency as the phase difference between the cantilever deflection signal and the driving signal. We adjusted the setpoint amplitude A1,set until we obtained a stable interaction phase Φ1 of below 60°. For this setting the ratio between set and free amplitude is around 0.7. A small amount of frequency shift Δf2 was applied via the piezoelectric actuator to maintain the second eigenmode on resonance as the tip interacts with the sample. To estimate the tip radius (R), we used reference samples (polystyrene with a modulus of 3 GPa69 and fused silica with a modulus of 73 GPa). The closer the modulus of the reference and test sample, the more accurate the result. For example, the modulus of b/c aggregates is typically overestimated when measured against a calibration sample of fused silica, because the modulus of b/c (1–2 GPa) differs significantly from that of fused silica. So, we calibrated b/c electrode with polystyrene film sample. We first performed an AM-FM measurement on the reference sample and extracted the tip radius value by fitting the measured frequency shift to the corresponding theoretical equation. After the measurement, we repeated the calibration on the reference samples. We only considered measurements where the difference in tip radius values before and after the sample measurement was less than 10%. The second resonance frequency (f2) is theoretically 6.267 times larger than the first resonance frequency (f1).70 However, the actual ratio deviates slightly due to tip mass, cantilever geometry, and material inhomogeneity.71 In our experiments, the second resonance frequency was around 5.7 times larger than the first resonance frequency. We used high-frequency cantilever holder during AM-FM measurement to ensure accuracy at these low amplitudes. We employed the sigma (σ) value to represent the histogram distribution and fitting error, which was calculated using the following formula:
. Upon approaching the sample, the interaction phase Φ1 was determined at the first resonance frequency as the phase difference between the cantilever deflection signal and the driving signal. We adjusted the setpoint amplitude A1,set until we obtained a stable interaction phase Φ1 of below 60°. For this setting the ratio between set and free amplitude is around 0.7. A small amount of frequency shift Δf2 was applied via the piezoelectric actuator to maintain the second eigenmode on resonance as the tip interacts with the sample. To estimate the tip radius (R), we used reference samples (polystyrene with a modulus of 3 GPa69 and fused silica with a modulus of 73 GPa). The closer the modulus of the reference and test sample, the more accurate the result. For example, the modulus of b/c aggregates is typically overestimated when measured against a calibration sample of fused silica, because the modulus of b/c (1–2 GPa) differs significantly from that of fused silica. So, we calibrated b/c electrode with polystyrene film sample. We first performed an AM-FM measurement on the reference sample and extracted the tip radius value by fitting the measured frequency shift to the corresponding theoretical equation. After the measurement, we repeated the calibration on the reference samples. We only considered measurements where the difference in tip radius values before and after the sample measurement was less than 10%. The second resonance frequency (f2) is theoretically 6.267 times larger than the first resonance frequency (f1).70 However, the actual ratio deviates slightly due to tip mass, cantilever geometry, and material inhomogeneity.71 In our experiments, the second resonance frequency was around 5.7 times larger than the first resonance frequency. We used high-frequency cantilever holder during AM-FM measurement to ensure accuracy at these low amplitudes. We employed the sigma (σ) value to represent the histogram distribution and fitting error, which was calculated using the following formula:Here w denotes the full width at half maximum (FWHM) parameter extracted from the Gaussian fitting results. We also calculated the sample standard deviation (SD) to quantify the variability of the moduli across different crystal facets in summary of the calibrated modulus table (Table 1).

We used silica microspheres with a radius of ≈1.5 µm as calibration samples. Silica is mechanically isotropic, with a nominal modulus of 70 GPa.73 The measured apparent modulus reduces with increasing tilt angle (Fig. 6a). For tilt angles >≈70° the tip may slide sideways resulting in an undefined tip sample contact. Then modulus calculation becomes unreliable. The software detects these events and automatically masks the corresponding pixels during measurement. By extracting modulus values at different tilt angles, we obtained the distribution of measured modulus as a function of angle (Fig. 6b). The modulus initially decreases with increasing tilt angle, and then gradually increases beyond approximately 60°. To calculate modulus of tilted surfaces, we define a correction factor as the ratio between the apparent and nominal modulus of the silica sphere (Fig. 6c). A. N. Ahmine et al. reported that for a spherical tip, the correction factor scales with (1/cosθ)2.5, where θ is surface tilt angle.74 In our calibration, the decrease in apparent modulus from 0° to around 50° follows a (1/cosθ)2.5 dependence. In order to calculate the modulus of surfaces at any angle and to take into account non-spherical tip shapes, we used polynomial fitting to derive a specific correction factor for our system (Fig. 6c). The fitting equation was:
| Kc = 1.00031 + 0.00161θ − 1.43861 × 10−4θ2 − 2.55867 × 10−5θ3 + 4.53632 × 10−7θ4 |
θ)n dependence, and the polynomial fit aligns well with the raw data with an applied fitting cut-off angle of 45°. In all subsequent data analysis, we apply this equation to compute the correction factor and mask regions with local tilt angles above 45°.
 | ||
| Fig. 6 Modulus calibration for different local tilt angles. (a) 3D topography of silica microspheres. The overlayed color scale represents the apparent modulus. (b) Apparent modulus in dependence on surface tilt angle. The error bars correspond to the standard deviation of the mean. (c) The ratio of apparent to real modulus as a function of surface tilt angle (θ). The blue line represents a polynomial fit from 0 to 45°. The red line shows a correction factor based on a geometrical relationship (1/cosθ)2.5. | ||
The CasaXPS software was used for spectral analysis, employing a Shirley background and employing the Gaussian–Lorentzian GL (30) peak shape. Energy calibration was performed using the peak for sp3 carbon (C–C) from the surface layer hydrocarbon species at 284.8 eV. The calibration was verified by checking the binding energy positions for spectra of all other elements. The probing depth for the measurements is estimated at approximately 14 nm, using polyethylene as a material representative in density to the interphase layer, and employing the TPP-M2 equation75,76 (probing depth ≈ 3 times the inelastic mean free path of electrons). The binding energy and assignment of each peak are presented in the Table S1.
000 counts per second. Charge compensation was maintained using a low-energy electron flood gun. An analysis area of 25 × 25 µm2 was rastered at a 512 × 512 pixel resolution with a 100 µs cycle time (mass range m/z = 0–963 u). Spectra were calibrated using O−, C2−, MnO2− and NiO2− fragments. Given the nanometer-thick CEI, the primary ion dose during image acquisition was sufficient to provide depth resolved information. Since sputter yields were unknown, ion fluence was used to represent depth resolved information.
Data evaluation was conducted with SurfaceLab 7.6 software (IONTOF, Germany). Regions of Interest for the LNMO particles and b/c region were defined using intensity thresholds of NiO2− and C2− signals, respectively. This segmentation allows for a comparison of the CEI chemical composition across the different electrodes.
Author contributions
X. S. carried out the batteries testing and SFM experiments, performed corresponding data collection and analysis. A. N. performed XPS experiment and corresponding data analysis. T. F. and S. S. performed TOF-SIMS experiments and corresponding data analysis with A. H. X. S. wrote the manuscript. F. W., H.-J. B. and R. B. provided expertise, SFM input, feedback, and revised the manuscript. A. N., T. F. and S. S. provided expertise, composition analysis, feedback, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are openly available on the Edmond cloud repository at the following link: https://doi.org/10.17617/3.67LFKI.Supplementary information (SI): SEM and EDX measurement, SFM characterizations, cycling performance data, standard nanoindentation measurements, ToF-SIMS analysis, impedance measurements and XPS spectra. See DOI: https://doi.org/10.1039/d5ta09458e.
Acknowledgements
X. S. acknowledges support from CSC (contract 202204910093). X. S. and R. B. acknowledges João Paulo Cosas Fernandes (University of Grenoble Alpes) for inspiring the application of the AM-FM technique and for valuable discussions. X. S. acknowledges Gabriele Selvaggio, Florian Johann and Marta Kocun (Oxford Instruments Company) for helpful discussions regarding AM-FM method. We thank Chao Zhu (MPI-P) and Liying Shen (MPI-P) for discussions on the experimental work, Helma Burg (MPI-P) and Leonard Ries (MPI-P) for laboratory assistance, Gunnar Glasser (MPI-P) and Ingo Lieberwirth (MPI-P) for support with electron microscopy characterization, Maren Müller (MPI-P) for focused ion beam measurement, Michael Kappl (MPI-P) for discussion on nanoindentation experiments, Robin Elo (Uppsala university) for the support with electron microscopy characterization. F. W. acknowledges support from the Max Planck Graduate Center with the Johannes Gutenberg University (MPGC). Open Access funding provided by the Max Planck Society.References
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- J. L. Li, Y. N. Wang, S. Y. Sun, Z. Zheng, Y. Gao, P. Shi, Y. J. Zhao, X. Li, Q. Li, X. Q. Zhang and J. Q. Huang, Adv. Energy Mater., 2024, 15, 2403845 CrossRef.
- S. Kuppan, Y. H. Xu, Y. J. Liu and G. Y. Chen, Nat. Commun., 2017, 8, 14309 CrossRef CAS PubMed.
- Q. Deng, R. Hu, C. Xu, B. Chen and J. Zhou, J. Solid State Electrochem., 2019, 23, 2999–3008 CrossRef CAS.
- Y. Qin, K. Xu, Q. Wang, M. Ge, T. Cheng, M. Liu, H. Cheng, Y. Hu, C. Shen, D. Wang, Y. Liu and B. Guo, Nano Energy, 2022, 96, 107082 CrossRef CAS.
- H. Tavassol, M. K. Y. Chan, M. G. Catarello, J. Greeley, D. G. Cahill and A. A. Gewirth, J. Electrochem. Soc., 2013, 160, A888–A896 CrossRef CAS.
- W. H. Hou, Q. Feng, C. Liu, X. Zhang, J. Yue, Q. Tian, S. Wu, Y. Ou, P. Zhou, Y. Xia, Y. Wang, X. Song, H. Zhou, Y. Lu, S. Yan and K. Liu, Adv. Mater., 2025, 37, e2503893 CrossRef PubMed.
- H. Shin, J. Park, S. Han, A. M. Sastry and W. Lu, J. Power Sources, 2015, 277, 169–179 CrossRef CAS.
- J. Xu, Nano-Micro Lett., 2022, 14, 166 CrossRef CAS.
- P. Bai, X. Ji, J. Zhang, W. Zhang, S. Hou, H. Su, M. Li, T. Deng, L. Cao, S. Liu, X. He, Y. Xu and C. Wang, Angew Chem., Int. Ed. Engl., 2022, 61, e202202731 CrossRef CAS PubMed.
- J. Huang, J. Liu, J. He, M. Wu, S. Qi, H. Wang, F. Li and J. Ma, Angew Chem., Int. Ed. Engl., 2021, 60, 20717–20722 CrossRef CAS PubMed.
- Y. Jiang, Y. Lu, Z. Zhang, L. Chang, J. Li, X. Han, L. Gan, Y. Ni, M. Sui and P. Yan, Mater. Res. Lett., 2023, 11, 471–479 CrossRef CAS.
- Y. K. Lee, J. Power Sources, 2021, 484, 229270 CrossRef CAS.
- P. Stüble, V. Mereacre, H. Geßwein and J. R. Binder, Adv. Energy Mater., 2023, 13, 2203778 CrossRef.
- J. Liu, J. Wang, Y. Ni, J. Liu, Y. Zhang, Y. Lu, Z. Yan, K. Zhang, Q. Zhao, F. Cheng and J. Chen, Angew Chem., Int. Ed. Engl., 2022, 61, e202207000 CrossRef CAS PubMed.
- Y. Gao and B. Zhang, Adv. Mater., 2023, 35, 2205421 CrossRef CAS.
- E. Darnbrough, J. Aspinall, M. Pasta and D. E. J. Armstrong, Mater. Des., 2023, 233, 112200 CrossRef CAS.
- S. K. Pradhan, B. B. Nayak, S. S. Sahay and B. K. Mishra, Carbon, 2009, 47, 2290–2292 CrossRef CAS.
- L. Ma, L. Levine, R. Dixson, D. Smith and D. Bahr, in Nanoindentation in Materials Science, 2012, ch. 2, DOI:10.5772/48106.
- A. Tiwari, Nanomechanical Analysis of High Performance Materials, Springer, 2014 Search PubMed.
- S. Daboss, N. Franke, B. Fraboni, C. Kranz and T. Cramer, J. Microsc., 2025, 1–12 Search PubMed.
- E. D. Hintsala, U. Hangen and D. D. Stauffer, Jom, 2018, 70, 494–503 CrossRef.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q. H. Yang, M. Wagemaker, F. Kang, X. Q. Yang and B. Li, Adv. Mater., 2019, 31, e1806620 CrossRef.
- Q. D. Nguyen, E.-S. Oh and K.-H. Chung, Polym. Test., 2019, 76, 245–253 CrossRef CAS.
- R. Garcia, Chem. Soc. Rev., 2020, 49, 5850–5884 RSC.
- N. Raßmann, S. Trippmacher, A. Specht, K. Theis, T. Rößler, S. Wohlrab, R. Kellnberger, S. Salehi, H. Bargel, N. Helfricht, K. Albrecht, T. Scheibel, J. Groll, S. Gekle and G. Papastavrou, ACS Appl. Nano Mater., 2025, 8, 5383–5398 CrossRef.
- R. Garcia and R. Proksch, Eur. Polym. J., 2013, 49, 1897–1906 CrossRef CAS.
- Z. Zhang, K. Smith, R. Jervis, P. R. Shearing, T. S. Miller and D. J. L. Brett, ACS Appl. Mater. Interfaces, 2020, 12, 35132–35141 CrossRef CAS.
- J. Zhang, X. Yang, R. Wang, W. Dong, W. Lu, X. Wu, X. Wang, H. Li and L. Chen, J. Phys. Chem. C, 2014, 118, 20756–20762 CrossRef CAS.
- Y. Gu, W. W. Wang, Y. J. Li, Q. H. Wu, S. Tang, J. W. Yan, M. S. Zheng, D. Y. Wu, C. H. Fan, W. Q. Hu, Z. B. Chen, Y. Fang, Q. H. Zhang, Q. F. Dong and B. W. Mao, Nat. Commun., 2018, 9, 1339 CrossRef PubMed.
- S. Wang, X. Yin, D. Liu, Y. Liu, X. Qin, W. Wang, R. Zhao, X. Zeng and B. Li, J. Mater. Chem. A, 2020, 8, 18348–18357 RSC.
- C. Gamonpilas and E. P. Busso, Mater. Sci. Eng., A, 2004, 380, 52–61 CrossRef.
- J. Degenhardt, X. Hu, M. Zackaria, F. Menelao, R. Tutsch and G. Dai, Nanomanuf. Metrol., 2025, 8, 15 CrossRef.
- R. Bolin, H. Yavas, H. Song, K. J. Hemker and S. Papanikolaou, Crystals, 2019, 9, 652 CrossRef CAS.
- M. Kocun, A. Labuda, W. Meinhold, I. Revenko and R. Proksch, ACS Nano, 2017, 11, 10097–10105 CrossRef CAS PubMed.
- A. Labuda, M. Kocun, W. Meinhold, D. Walters and R. Proksch, Beilstein J. Nanotechnol., 2016, 7, 970–982 CrossRef CAS.
- H.-J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- M. S. Kashani and V. Madhavan, Acta Mater., 2011, 59, 883–895 CrossRef CAS.
- M. T. Nguyen, H. Q. Pham, J. A. Berrocal, I. Gunkel and U. Steiner, J. Mater. Chem. A, 2023, 11, 7670–7678 RSC.
- F. P. McGrogan, Y.-M. Chiang and K. J. Van Vliet, J. Electroceram., 2016, 38, 215–221 CrossRef.
- N. Sharma, D. Meng, X. Wu, L. S. de Vasconcelos, L. Li and K. Zhao, Extreme Mech. Lett., 2023, 58, 101920 CrossRef.
- L. Wu and J. Zhang, J. Appl. Phys., 2015, 118, 225101 CrossRef.
- J. Zhang, O. Cohen, X. Lang, B. Wang, S. E. Trask, K. Cho, K. A. Persson and L. Su, Small, 2025, 21, e2502141 CrossRef.
- A. Manthiram, K. Chemelewski and E.-S. Lee, Energy Environ. Sci., 2014, 7, 1339–1350 RSC.
- A. H. Alsaedi, B. F. Spencer, S. Sheraz, A. S. Walton and N. P. Lockyer, ACS Omega, 2025, 10, 57607–57622 CrossRef CAS PubMed.
- Z. Zhang, J. Yang, W. Huang, H. Wang, W. Zhou, Y. Li, Y. Li, J. Xu, W. Huang, W. Chiu and Y. Cui, Matter, 2021, 4, 302–312 CrossRef CAS.
- R. R. Liu, X. Deng, X. R. Liu, H. J. Yan, A. M. Cao and D. Wang, Chem. Commun., 2014, 50, 15756–15759 RSC.
- L. Wheatcroft, N. Klingner, R. Heller, G. Hlawacek, D. Ozkaya, J. Cookson and B. J. Inkson, ACS Appl. Energy Mater., 2020, 3, 8822–8832 CrossRef CAS PubMed.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef.
- J. C. Hestenes, J. T. Sadowski, R. May and L. E. Marbella, ACS Mater. Au, 2023, 3, 88–101 CrossRef CAS PubMed.
- R. Dedryvère, D. Foix, S. Franger, S. Patoux, L. Daniel and D. Gonbeau, J. Phys. Chem. C, 2010, 114, 10999–11008 CrossRef.
- H. Chen, A.-M. Mattsson, L. King, H. Liu, I. Nielsen, T. Ericson, A. Preobrajenski, W. R. Brant and M. Hahlin, J. Mater. Chem. A, 2024, 12, 25393–25406 RSC.
- S. R. Daemi, C. Tan, T. Volkenandt, S. J. Cooper, A. Palacios-Padros, J. Cookson, D. J. L. Brett and P. R. Shearing, ACS Appl. Energy Mater., 2018, 1, 3702–3710 CrossRef CAS.
- D. Cao, C. Tan and Y. Chen, Nat. Commun., 2022, 13, 4908 CrossRef CAS PubMed.
- J.-N. Zhang, Q. Li, Y. Wang, J. Zheng, X. Yu and H. Li, Energy Storage Mater., 2018, 14, 1–7 CrossRef.
- W. Lu, J. Zhang, J. Xu, X. Wu and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 19313–19318 CrossRef CAS PubMed.
- J. Alvarado, M. A. Schroeder, T. P. Pollard, X. Wang, J. Z. Lee, M. Zhang, T. Wynn, M. Ding, O. Borodin, Y. S. Meng and K. Xu, Energy Environ. Sci., 2019, 12, 780–794 RSC.
- Z. Zhao, W. Zhang, D. Wang, L. Li, Q. Liang, W. Li, C. Lu, S. Jo Yoo, J. G. Kim, Z. Chen, Y. Li, X. Zou, F. Liu, X. Zhou, K. Song, J. Li and W. Zheng, Angew Chem., Int. Ed. Engl., 2024, 63, e202414420 CrossRef CAS PubMed.
- M. Steinhauer, S. Risse, N. Wagner and K. A. Friedrich, Electrochim. Acta, 2017, 228, 652–658 CrossRef CAS.
- M. D. Bouguern, A. K. M R and K. Zaghib, J. Power Sources, 2024, 623 CrossRef CAS.
- T. Kim, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2023, 11, 221–231 RSC.
- J. Maibach, F. Lindgren, H. Eriksson, K. Edstrom and M. Hahlin, J. Phys. Chem. Lett., 2016, 7, 1775–1780 CrossRef CAS.
- J. Tan, J. Matz, P. Dong, J. Shen and M. Ye, Adv. Energy Mater., 2021, 11, 2100046 CrossRef CAS.
- K. Tasaki, A. Goldberg, J.-J. Lian, M. Walker, A. Timmons and S. J. Harris, J. Electrochem. Soc., 2009, 156, A1019 CrossRef CAS.
- C. Weng, M. Qiu, B. Wang, J. Yang, W. Mai, L. Pan, S. Huang and J. Li, Angew Chem., Int. Ed. Engl., 2025, 64, e202419539 CrossRef CAS PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- J. W. Li, J. P. Cleveland and R. Proksch, Appl. Phys. Lett., 2009, 94, 163118 CrossRef.
- G. Chawla and S. D. Solares, Appl. Phys. Lett., 2011, 99, 074103 CrossRef.
- E. Bonaccurso, B. Cappella and K. Graf, J. Phys. Chem. B, 2006, 110, 17918–17924 CrossRef CAS.
- B. Eslami and M. Damircheli, J. Appl. Phys., 2019, 126, 095301 CrossRef.
- M. Damircheli and B. Eslami, Beilstein J. Nanotechnol., 2020, 11, 1525–1541 CrossRef CAS PubMed.
- M. Laurent-Brocq, E. Bejanin and Y. Champion, Scanning, 2015, 37, 350–360 CrossRef PubMed.
- S. Bruns, K. E. Johanns, H. U. R. Rehman, G. M. Pharr and K. Durst, J. Am. Ceram. Soc., 2017, 100, 1928–1940 CrossRef CAS.
- A. N. Ahmine, M. Bdiri, S. Fereol and R. Fodil, Sci. Rep., 2024, 14, 25869 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2004, 21, 165–176 CrossRef.
- C. J. Powell and A. Jablonski, J. Surf. Anal., 2002, 9, 322–325 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |