
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Free energy relationship analysis for temperature dependence of hydride kinetic isotope effects of NADH/NAD+ model reactions: implication for barrier compression by enzyme dynamics
Ava Austin-Kloppe†
,
Nicholas DeGroot†,
Bikram Dhakal,
Jessica Sager,
Lauren Phan,
Seyedmehrad Poormoghim and
Yun Lu *
*
Department of Chemistry, Southern Illinois University Edwardsville, Edwardsville, Illinois 62026, USA. E-mail: yulu@siue.edu
First published on 6th May 2026
Abstract
The observed shift from temperature (T)-independence of hydrogen kinetic isotope effects (KIEs) in wild-type enzymes to T-dependence of KIEs in enzyme mutants has been explained as evidence for the role of protein dynamics in compressing donor (Don)–acceptor (Acc) distances (DADs) for catalysis. To test this explanation, correlation analysis of free energy changes (ΔG° = −44.3 to 6.7 kcal mol−1) that simulate system rigidities and T-dependence of KIEs (represented by ΔEa = EaD − EaH) was carried out for 34 hydride-tunneling reactions of NADH/NAD+ models in acetonitrile. For exergonic reactions, ΔEa increases as ΔG° approaches zero, with the linear trend appearing to reverse for endergonic reactions. Both ΔEa and KIEs reach their maximum near thermoneutral reactions, where the charge-transfer (CT) complexation vibration is the weakest and DAD is the longest. A small portion of the free energy change drives the CT complexation vibrations and thus the DAD sampling that correlates with KIEs and their T-dependences. The results support the role of protein dynamics in barrier compression for catalysis. The new physical-organic linear ΔEa–ΔG° relationship will contribute to the development of future H-tunneling models as well as updated theories for enzyme catalysis.
Introduction
Recent observations of temperature (T) independence (or weak dependence) of kinetic isotope effects (KIEs) in various enzyme-catalyzed hydrogen (H) transfer reactions have been ascribed by several research groups to the T-independence of narrowly distributed donor–acceptor distances (DADs) for H-tunneling processes.1–14 Therefore, such observations are often cited in support of the proposed role of strong protein vibrational dynamics in facilitating short DAD sampling,1,15–20 and further promoting enzyme catalysis.8,12,20–31 Use of this approach to propose the new physical origin of enzyme catalysis is currently, however, debated.32–36 Central to the debate is whether T-dependence of KIEs reliably reflects DAD fluctuations or overall system rigidity. To address this question, biochemists have designed experiments to probe enzyme system rigidity and its correlation with the T-dependence of KIEs.6,9,11,12,37–39 Theoreticians have refined or developed H-tunneling models to simulate the observations in attempts to test whether there is such a correlation.14,32,33,40–43 For the same purpose, our group has studied model reactions in solution to explore the potential relationship between the structure and T-dependence of hydride KIEs.24,25,27,29,44–50 The solution-phase H-transfer reactions permit systematic variation of the molecular structure and solvent environment, enabling more controlled evaluation of how structural rigidity influences the T-dependence of KIEs in a broader context.T-Dependence of KIEs reflects the isotopic activation energy difference, represented as ΔEa (= EaD − EaH) for hydrogen/deuterium transfer reactions. Semi-classically, ΔEa falls within the range between 1.0 and 1.2 kcal mol−1, but its relationship with structure is unpredictable. ΔEa outside of this range has been used to suggest an H-tunneling mechanism. Notably, a shift from ΔEa ∼0 in wild-type enzymes to ΔEa > 0 (often exceeding the semi-classical limit) in mutant variants has been frequently observed. This trend has prompted the application of existing H-tunneling models, as well as the development of new theoretical frameworks, to account for these observations and to further elucidate the mechanisms of H-transfer chemistry.2,5–9,11,12,18,31,42,51–58 Among these, the recently proposed vibration-assisted activated H-tunneling (VA-AHT) model appears to be able to explain the unusual KIE behaviors.2,5,8,20,59
The VA-AHT model incorporates two orthogonal activation processes: (1) heavy atom motions bring the reactants (donor-H and acceptor) and products to degenerate energy states at which H-wave functions overlap ([Don-H ↔ H-Acc]‡), i.e., tunnelling ready states (TRS's); and (2) more rapid heavy atom motions, also called promoting vibrations,19,60 sample short DADs for efficient tunneling to occur. Since tunneling of a D-isotope requires a smaller average DAD due to its shorter de Broglie wavelength, a higher activation energy is needed leading to EaD > EaH (assuming that the first activation process is isotope insensitive). In wild-type enzymes, however, strong protein vibrations compress the donor/acceptor closely together, thereby facilitating the formation of short DADs that are extremely densely populated, eliminating the possibility for further short DAD sampling for D-tunneling and making ΔEa ∼0. In enzyme mutants, the constructive vibrations are disrupted, and thermal sampling of shorter DADs is allowed so that T-dependence of DADs/KIEs (ΔEa > 0) emerges. The phenomenological model has been claimed by some researchers to be able to explain all of the hydrogen KIEs.5,6,8,12
Computational simulations of T-dependence of KIEs followed various theoretical models including the VA-AHT model to investigate the proposed role of fast thermal dynamics in sampling short DADs. Other models include ensemble-averaged variational TS theory with multi-dimensional H-tunneling32,41,61 as well as approaches using the empirical valence bond theory.33,34,62 While the VA-AHT model has successfully reproduced the ΔEa in nonadiabatic reactions to support the thermally activated DAD sampling concept,63–65 simulations for more adiabatic hydride/proton transfer reactions have been challenging as there is no direct mathematical relationship yet between DADs and ΔEa for this type of reaction.30,43 On the other hand, theoretical replications of the observations using other H-tunneling models, especially the huge KIEs, have encountered difficulty.10,32,36,66 Nevertheless, even some of the latter computational results sometimes show that ΔEa ∼ 0 in some reactions results from the insensitivity of the DAD to temperature,13,34,41,63 whereas other researchers have shown that it could also result from the effect of temperature on the microscopic mechanism, for example, on the position of the TS and shape of the potential barrier.32,34 The potential relationship between DAD distributions and ΔEa magnitudes warrants further investigation to search for a potential mathematical relationship, if it exists, for building future H-tunnelling theoretical frameworks.
To address the “DAD–ΔEa” relationship, additional data, including from model systems, are needed. Over the years, we have designed hydride transfer reactions of NADH/NAD+ models in solution to tackle the problem. Our hypothesis, based on enzyme observations, is that a more rigid system with densely populated DADs gives rise to a smaller ΔEa.25 Systems have been designed to study the electronic, steric, and solvent effects as well as effects of remote heavy group vibrations and mechanisms on the T-dependence of KIEs.25,27,29,44–50 Current results show that a tighter charge-transfer (CT) complexation between NADH/NAD+ model structures exhibits a smaller ΔEa value, supporting the hypothesis.
As the project progressed, we seemed to have identified a trend indicating that a reaction with a more negative free energy change (ΔG°) tends to show a smaller ΔEa value.29,44,46,48 This appears consistent with our hypothesis as a stronger hydride donor/acceptor would form stronger CT complexation vibrations and thus more rigid donor–acceptor centers. In this context, a larger negative ΔG° corresponds to a tighter CT complexation and, consequently, more densely populated smaller DADs, which in turn result in smaller ΔEa values. Based on this reasoning, we envisioned that there might be a free energy relationship with T-dependence of KIEs for this type of reaction. As a matter of fact, a VA-AHT-inspired model can be formulated in which a portion of the free energy  drives the structural and solvent rearrangement required to reach a TRS, while the remaining portion
drives the structural and solvent rearrangement required to reach a TRS, while the remaining portion  modulates DAD sampling toward shorter distances; together, both contributions enable H-tunneling. While the overall free energy change
modulates DAD sampling toward shorter distances; together, both contributions enable H-tunneling. While the overall free energy change  is expected to linearly correlate with the logarithm of the observed rates (ln(k)), ΔEa reflects (but is not equal to) the thermal energy required for DAD sampling and is expected to correlate linearly with the
is expected to linearly correlate with the logarithm of the observed rates (ln(k)), ΔEa reflects (but is not equal to) the thermal energy required for DAD sampling and is expected to correlate linearly with the  . Although
. Although  cannot be directly determined, establishing a ΔEa–ΔG° relationship would provide an indirect means to test our DAD–ΔEa hypothesis, clarify the driving force underlying ΔEa, and deepen our understanding of DAD sampling as well as the enzymatic KIE behavior.
cannot be directly determined, establishing a ΔEa–ΔG° relationship would provide an indirect means to test our DAD–ΔEa hypothesis, clarify the driving force underlying ΔEa, and deepen our understanding of DAD sampling as well as the enzymatic KIE behavior.
In this work, we examined 34 hydride transfer reactions of NADH/NAD+ models in acetonitrile, with ΔG° values spanning from −44.3 to 6.7 kcal mol−1, to investigate the previously unreported ΔEa–ΔG° relationship and further test our hypothesis concerning the DAD–ΔEa relationship. We also compare the ΔEa–ΔG° relationship with the ln(k)–ΔG° and ln(KIE)–ΔG° relationships. Furthermore, this study represents the first application of our hypothesis to endergonic reactions. The dataset reveals how the trends of these relationships evolve when transitioning from exergonic to endergonic regions. These insights enable testing of the DAD sampling mechanism in the VA-AHT(-inspired) model, evaluating other current H-transfer theories, as well as informing the development of future theoretical frameworks for both general H-transfer/tunneling reactions and enzyme catalysis.
Results
The hydride donor and acceptor structures are shown in Fig. 1. Kinetics were determined spectroscopically on a stopped-flow UV-vis spectrophotometer. The ΔG°, second-order rate constants (k2) and KIEs at 25 °C, as well as the ΔEa values are listed in Table 1 (for Rxns 1–34). As noted, some data were taken from our previous publications. Although kinetic study of the endergonic reactions is challenging due to the limited extent of the reaction and slow kinetics, in this work we were able to conduct a study on two such hydride transfer reactions: from Hantzsch 1,4-dihydropyridine (HEH) to benzylidenemalononitriles (BMNs), NBMN (ΔG° = 6.3 kcal mol−1) and TBMN (6.7 kcal mol−1) (Rxns 33 and 34 in Table 1; see structures in Fig. 1). The reactions proceed to completion due to subsequent rapid irreversible transfer of a proton from the N–H site of the HE+ product to the dicyanomethanide anion product, as illustrated below,67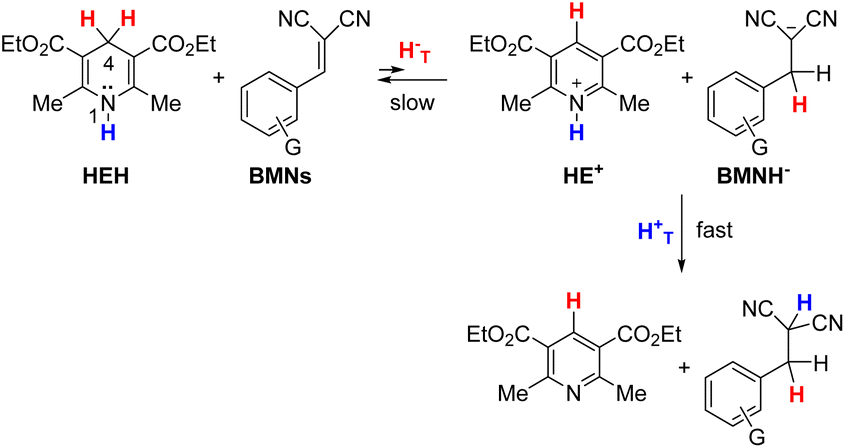
 | ||
| Fig. 1 Hydride donors and acceptors (counter-ions: BF4−) (L = H or D) of the reactions in acetonitrile. | ||
 , free energy changes (ΔG°), and hydride transfer kinetic data in acetonitrile
, free energy changes (ΔG°), and hydride transfer kinetic data in acetonitrile
| Rxns | Donors (Don-H) | Hydride acceptors (Acc+BF4−) |
|
|
ΔG° (kcal mol−1) | k2H (25 °C)l (M−1 s−1) | KIEm (25 °C) | ΔEam (kcal mol−1) |
|---|---|---|---|---|---|---|---|---|
| a Ref. 44.b Ref. 25.c This work.d ΔEa = 1.53 (0.15) was reported by us before.25 This was determined with temperatures from 4.5–29.5 °C, using the Hi-Tech Scientific SFA-20 fast kinetic determination kit interfaced to a UV-vis spectrophotometer. The conclusions from that publication are not changed using the numbers obtained in this work.e Ref. 48.f Ref. 29.g Ref. 47.h Ref. 24.i Ref. 29.j Unreported.k For 22 °C.l Ref. 68.m Numbers in parentheses are pooled standard deviations (standard deviations of the average values from different days of measurements in this work are listed in Tables S1 to S10 for comparison). | ||||||||
| Exergonic reactions | ||||||||
| 1a | DMPBIH | CF3PhXn+ | 49.2 | 93.5 | −44.3 | 1.44 (0.01) × 105 | 2.56 (0.03) | 0.03 (0.07) |
| 2a | DMPBIH | BrPhXn+ | 49.2 | 92.5 | −43.3 | 9.86 (0.09) × 104 | 2.55 (0.03) | 0.07 (0.07) |
| 3a | DMPBIH | PhXn+ | 49.2 | 91.6 | −42.4 | 4.54 (0.05) × 104 | 2.68 (0.04) | 0.27 (0.06) |
| 4a | DMPBIH | CH3OPhXn+ | 49.2 | 90.2 | −41.0 | 2.08 (0.02) × 104 | 2.74 (0.03) | 0.55 (0.06) |
| 5a | DMPBIH | (CH3)2NPhXn+ | 49.2 | 86.7 | −37.5 | 6.34 (0.04) × 102 | 2.89 (0.06) | 0.50 (0.08) |
| 6b | DMPBIH | MA+ | 49.2 | 76.2 | −27.0 | 2.12 (0.01) × 102 | 3.57 (0.03) | 0.43 (0.15) |
| 7a | MAH | CF3PhXn+ | 76.2 | 93.5 | −17.3 | 1.03 (0.01) × 103 | 4.06 (0.04) | 0.89 (0.07) |
| 8a | MAH | BrPhXn+ | 76.2 | 92.5 | −16.3 | 6.45 (0.03) × 102 | 4.04 (0.03) | 0.89 (0.07) |
| 9a | MAH | PhXn+ | 76.2 | 91.6 | −15.4 | 4.10 (0.03) × 102 | 4.08 (0.03) | 0.88 (0.05) |
| 10a | MAH | CH3OPhXn+ | 76.2 | 90.2 | −14.0 | 1.56 (0.01) × 102 | 4.18 (0.04) | 0.92 (0.16) |
| 11a | MAH | (CH3)2NPhXn+ | 76.2 | 86.7 | −10.5 | 4.34(0.03) | 4.45 (0.05) | 0.96 (0.18) |
| 12c | MAH | Tr+ | 76.2 | 83.0 | −6.8 | 4.01 (0.02) | 4.95 (0.12) | 1.14 (0.10) |
| 13c | HAH | Tr+ | 74.9 | 83.0 | −8.1 | 7.33 (0.05) × 10 | 5.34 (0.04) | 1.27 (0.09) |
| 14c | HAH | PhXn+ | 74.9 | 91.6 | −16.7 | 1.15 (0.01) × 103 | 4.19 (0.03) | 0.88 (0.05) |
| 15e | BAH | PhXn+ | 77.4 | 91.6 | −14.2 | 3.79 (0.02) × 102 | 4.26 (0.03) | 0.89 (0.05) |
| 16e | MPH | PhXn+ | 65.7 | 91.6 | −25.9 | 3.74 (0.02) × 103 | 3.18 (0.03) | 0.71 (0.05) |
| 17c | MPH | CH3OPhXn+ | 65.7 | 90.2 | −24.5 | 1.65 (0.01) × 103 | 3.28 (0.03) | 0.63 (0.05) |
| 18c,d | BNAH | MA+ | 59.3 | 76.2 | −16.9 | 7.16 (0.07) × 10 | 4.59 (0.06) | 1.14 (0.17) |
| 19c | BNAH | BA+ | 59.3 | 77.4 | −18.1 | 2.48 (0.02) × 102 | 3.75 (0.03) | 0.82 (0.07) |
| 20c | BNAH | (CH3OPh)3C+ | 59.3 | 88.6 | −29.3 | 1.66 (0.01) × 105 | 2.80 (0.03) | 0.75 (0.05) |
| 21e | BNAH | (CH3)2NPhXn+ | 59.3 | 86.7 | −27.4 | 5.62 (0.04) × 104 | 3.19 (0.03) | 0.82 (0.04) |
| 22e | BNAH | (CH3)2NPhMA+ | 59.3 | 67.4 | −8.1 | 8.37 (0.08) × 10−1 | 4.79 (0.06) | 1.14 (0.21) |
| 23b | HEH | MA+ | 64.4 | 76.2 | −11.8 | 1.56 (0.01) × 102 | 4.92 (0.04) | 0.99 (0.11) |
| 24f | HEH | BA+ | 64.4 | 77.4 | −13.0 | 5.73 (0.03) × 102 | 4.53 (0.04) | 1.01 (0.12) |
| 25c | HEH | (CH3OPh)3C+ | 64.4 | 88.6 | −24.2 | 5.66 (0.03) × 103 | 3.64 (0.03) | 0.88 (0.06) |
| 26g | HEH | CF3PhMA+ | 64.4 | 78.4 | −14.0 | 2.74 (0.03) × 10 | 5.23 (0.05) | 1.13 (0.19) |
| 27g | HEH | BrPhMA+ | 64.4 | 75.8 | −11.4 | 2.09 (0.03) × 10 | 5.20 (0.08) | 1.32 (0.09) |
| 28g | HEH | PhMA+ | 64.4 | 74.1 | −9.7 | 1.37 (0.08) × 10 | 5.31 (0.04) | 1.19 (0.07) |
| 29g | HEH | CH3PhMA+ | 64.4 | 72.8 | −8.4 | 1.13 (0.02) × 10 | 5.30 (0.10) | 1.33 (0.10) |
| 30g | HEH | CH3OPhMA+ | 64.4 | 71.7 | −7.3 | 1.00 (0.01) × 10 | 5.11 (0.08) | 1.31 (0.10) |
| 31g | HEH | (CH3)2NPhMA+ | 64.4 | 67.4 | −3.0 | 4.19 (0.03) | 5.09 (0.06) | 1.27 (0.14) |
| 32g | HEH | (CH3)2NPhXn+ | 64.4 | 86.7 | −22.3 | 8.87 (0.05) × 104 | 3.56 (0.02) | 0.86 (0.08) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Endergonic reactions | ||||||||
| 33c | HEH | NBMN | 64.4 | 58.1 | 6.3 | 1.14 (0.01) | 5.13 (0.06) | 1.17 (0.15) |
| 34c | HEH | TBMN | 64.4 | 57.7 | 6.7 | 6.07 (0.02) × 10−1 | 5.27 (0.06) | 1.19 (0.16) |
| 35h | BnOH | PhXn+ | —j | 91.6 | —j | 6.73 (0.29) × 10−5k | 4.83 (0.21)k | 1.00 (0.26) |
| 36i | i-PrOH | PhXn+ | —j | 91.6 | —j | 2.02 (0.05) × 10−5k | 3.63 (0.23)k | 0.83 (0.27) |
| 37i | i-PrOH-β,β-d6 | PhXn+ | —j | 91.6 | —j | 2.01 (0.05) × 10−5k | 3.64 (0.16)k | 0.80 (0.19) |
| 38i | cyclo-HexOH | PhXn+ | —j | 91.6 | —j | 2.68 (0.01) × 10−5k | 3.68 (0.16)k | 0.90 (0.27) |


Table 1 also summarizes the relevant kinetic results for our previous study of the unfavorable hydride transfer reactions from alcohols (R1CH(OH)R2) to PhXn+ in acetonitrile (Rxns 35–38).24,29
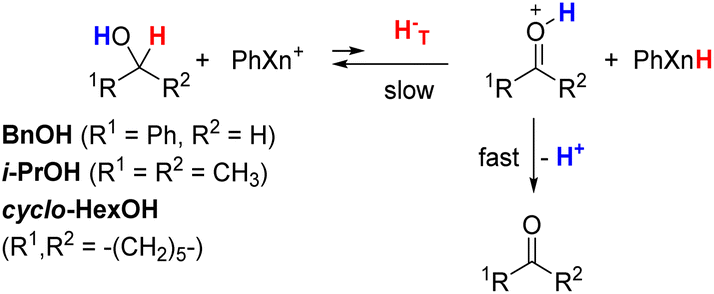
In these reactions, hydride-transfer from the α-position of the alcohol generates an α-hydroxy carbocation intermediate (R1C+(OH)R2). This process is subsequently followed by rapid proton transfer of the OH group to basic species in solution (e.g., excess alcohol substrate or acetonitrile solvent), producing the corresponding oxidized carbonyl product (R1C(O)R2).24,29,69–71
While these extremely slow hydride transfer processes are expected to be endergonic, the corresponding free energy data are not available in the literature, limiting quantitative free energy relationship analysis. Notably, these reactions proceed significantly more slowly than the endergonic reactions of HEH with BMNs (more than 104 times slower, see Table 1), suggesting that they would be even more endergonic. Estimated ΔG° values have therefore been obtained by extrapolating the linear ln(k2)–ΔG° correlation derived from reactions 1–34 (see the Discussion section below). These estimates are included to support the discussion of the trends observed across this limited set of endergonic reactions.
Discussion
Although the KIEs at 25 °C are all below 6, a majority of the ΔEa values fall outside of the semiclassical range from 1.0 to 1.2 kcal mol−1 (Table 1). These results are consistent with observations from enzyme-catalyzed hydride transfer reactions involving NADH/NAD+ coenzymes. In both enzymatic and solution systems, such findings have been analyzed to suggest tunneling mechanisms.5,48,72–74Free energy relationship analysis for T-dependence of KIEs: the Λ-shaped ΔEa–ΔG° correlation
We first performed a linear correlation analysis of all 34 ln(k2)–ΔG° data points (Fig. 2A). Although the fit is poor (R2 = 0.748), it reveals a clear trend that the reaction rate decreases as ΔG° becomes less negative within the exergonic region and continues to decrease as the reactions become endergonic. Analysis of the ΔEa–ΔG° relationship shows a relatively good linear correlation for the 32 exergonic reactions (Fig. 2B for Rxns 1–32, ΔG° = −44.3 to −3.0 kcal mol−1, R2 = 0.852), whereas the two endergonic reactions appear as significant outliers. Within the exergonic region, the ΔEa increases as ΔG° approaches zero. This new observation is consistent with our expectation; a more negative ΔG° corresponds to a more rigid system and a smaller ΔEa, with a portion of ΔG° contributing to the activation process associated with DAD sampling.
contributing to the activation process associated with DAD sampling.
 | ||
| Fig. 2 (A) The linear fit of the ln(k2H)–ΔG° data for the 34 hydride transfer reactions (Rxns 1–34). (B) The linear fit of the ΔEa–ΔG° data for the 32 exergonic hydride transfer reactions in acetonitrile (Rxns 1–32). The trend appears to turn to the opposite direction for endergonic reactions of BMNs (Rxns 33–34). Area between the two horizontal green dotted lines represents the semiclassical range. The reactions 35–38 are not included in both plots (see the text). | ||
Therefore, the two endergonic reactions appear to align with the 32 exergonic reactions in the linear ln(k2)–ΔG° correlation, whereas they appear as significant outliers relative to the linear ΔEa–ΔG° correlation for the 32 exergonic reactions. In the latter case, ΔEa reaches a maximum when the negative ΔG° approaches zero, but upon entering the endergonic region, it begins to decrease. One may think that this behavior resulted from the experimental error or from structural diversity among the reactants, where factors other than electronic effects, such as steric effects, can affect the CT complexation strength and, consequently, the DAD distributions. While we already restricted the structures to rings containing C, N, and O that involve only 2p orbitals in CT complexation and the reactions to only one-step hydride transfers between two carbons, to further minimize the structural variability factor, we replotted the ln(k2)–ΔG° and ΔEa–ΔG° correlations using only the 12 reactions with a common hydride donor, HEH (Fig. 3A and B, respectively). In both plots, the endergonic data points are clearly outliers.
 | ||
| Fig. 3 (A) The linear fit of the ln(k2H)–ΔG° data for the 12 hydride transfer reactions with a common HEH hydride donor (Rxns 23–34). (B) The linear fit of the ΔEa–ΔG° data for the same 12 exergonic hydride transfer reactions. In both plots, the trend appears to turn to the opposite direction for endergonic reactions 33–34. The empty green circle point in each plot is an extrapolated point at ΔG° = 0 derived from the linear equation from the exergonic reactions. | ||
As shown in Fig. 3A, the two endergonic reactions appear to turn the linear ln(k2)–ΔG° correlation derived from the exergonic reactions to the opposite direction. As shown in Fig. 3B, the two endergonic reactions clearly turn the linear ΔEa–ΔG° correlation for the exergonic reactions to the opposite direction. Using the extrapolated ΔG° = 0 point from the exergonic reaction data (the circled points in Fig. 3A and B), the corresponding linear correlations with the two data points from the endergonic reactions were drawn, which show a “V-shaped” ln(k2)–ΔG° relationship and a “Λ-shaped” ΔEa–ΔG° relationship.
We first discuss about the ΔEa–ΔG° relationship (Fig. 2B and 3B). Although the current analysis includes only two endergonic reaction data points, this trend could be reinforced by considering additional potential endergonic reactions 35–38. While the ΔG° values are not available for these reactions, a qualitative assessment can still be performed. These ΔG° values could be approximately derived by substituting the corresponding k2 values into the linear ln(k2)–ΔG° correlation equation for the reactions 1–34 (Fig. 2A). For one example, the k2 for the reaction of BnOH with PhXn+ in acetonitrile at 22 °C is 6.73 × 10−5 M−1 s−1 (Table 1). Using our reported rate data at other temperatures24 for an Arrhenius analysis, the rate constant at 25 °C is estimated to be 8.59 × 10−5 M−1 s−1. Substituting the number in the linear fit equation from Fig. 2A yields an estimated ΔG° = 43.4 kcal mol−1, indicating a highly endergonic process. We recognize that such an estimate could carry large uncertainty as the fit equation used depends upon many factors, such as dataset size, structural similarity among reactions, as well as experiment errors. Therefore, these estimated ΔG° values will not be used for quantitative analysis of the ΔEa–ΔG° relationship. Nevertheless, the ΔEa values for reactions 35–38 (0.80–1.00 kcal mol−1) are significantly lower than those of the two endergonic reactions (33–34; ∼1.2 kcal mol−1). These observations support the conclusion that the ΔEa–ΔG° trend for endergonic reactions is opposite to that observed for exergonic reactions.
The key question is why the clear linear ΔEa–ΔG° plot for the exergonic reactions reverses direction at ΔG° ∼ 0. This observation is reminiscent of the Marcus inverted region. In fact, the weakest CT complexation (i.e., the longest average DAD) at the TRS is expected from thermoneutral reactions  . This inference is based on Hammond's postulate. For an exergonic reaction, a more negative ΔG° would correspond with a tighter TRS that resembles more reactive reactant structures, whereas for an endergonic reaction, the more positive ΔG° would also correspond with a tighter TRS that, however, resembles more reactive products. Therefore, within the VA-AHT-inspired model,
. This inference is based on Hammond's postulate. For an exergonic reaction, a more negative ΔG° would correspond with a tighter TRS that resembles more reactive reactant structures, whereas for an endergonic reaction, the more positive ΔG° would also correspond with a tighter TRS that, however, resembles more reactive products. Therefore, within the VA-AHT-inspired model,  can be a proxy for TRS rigidity and a Λ-shaped ΔEa–ΔG° relationship is expected, with the maximum ΔEa occurring near ΔG° = 0 where the DAD is longest. Our experimental observations (Fig. 2B and considering the ΔEa values of the endergonic reactions 35–38) are largely consistent with this expectation. Therefore, not only the ΔEa–ΔG° correlation derived from exergonic reactions but the reversed ΔEa–ΔG° relationship for endergonic reactions also supports our proposed DAD–ΔEa relationship. That is, a more flexible system with a longer average DAD and a ΔG° closer to zero exhibits a larger ΔEa value, regardless of whether the reaction is exergonic or endergonic.
can be a proxy for TRS rigidity and a Λ-shaped ΔEa–ΔG° relationship is expected, with the maximum ΔEa occurring near ΔG° = 0 where the DAD is longest. Our experimental observations (Fig. 2B and considering the ΔEa values of the endergonic reactions 35–38) are largely consistent with this expectation. Therefore, not only the ΔEa–ΔG° correlation derived from exergonic reactions but the reversed ΔEa–ΔG° relationship for endergonic reactions also supports our proposed DAD–ΔEa relationship. That is, a more flexible system with a longer average DAD and a ΔG° closer to zero exhibits a larger ΔEa value, regardless of whether the reaction is exergonic or endergonic.
It should be noted that steric effects can influence the DAD distributions and, consequently, the ΔEa values. Increased steric crowding may enhance system rigidity, leading to a decrease in ΔEa.25 Conversely, steric hindrance may also physically separate the donor and acceptor, enhancing system flexibility and resulting in higher ΔEa values. Therefore, variations in steric effects across different systems may contribute to the observed scatter in the correlation, in addition to experimental errors.
The ln(k2)–ΔG° correlation is consistent with the Λ-shaped ΔEa–ΔG° relationship
Another question arises as to why the ΔEa values of the two endergonic reactions decrease (Fig. 3A) but their rates increase (Fig. 3B) as compared to their corresponding values at ΔG° = 0. The question can also be answered by considering the two orthogonal but coupled activation processes in the VA-AHT-inspired model, the TRS formation and DAD sampling. Note that TRS formation involves both structural (hybridization and charge) and solvation reorganizations. The observed k2 is determined by both processes (driven by both and
and  ) and can be expressed as k2 = (k2,TRS × k2,DAD)1/2 (or more generally, as a weighted geometric mean), where k2,TRS is the rate of reaching the TRS with correct donor–acceptor alignment and k2,DAD is the rate of reaching the TRS with short DADs; both structural features of the TRS are required for tunneling. Theoretically, ln(kTRS) would correlate linearly with the
) and can be expressed as k2 = (k2,TRS × k2,DAD)1/2 (or more generally, as a weighted geometric mean), where k2,TRS is the rate of reaching the TRS with correct donor–acceptor alignment and k2,DAD is the rate of reaching the TRS with short DADs; both structural features of the TRS are required for tunneling. Theoretically, ln(kTRS) would correlate linearly with the  across both the exergonic and endergonic regions, which is, ln(kTRS) decreases as
across both the exergonic and endergonic regions, which is, ln(kTRS) decreases as  increases from negative through zero to positive values (Fig. 4A). In contrast, ln(kDAD) is expected to decrease as exergonic reactions approach thermoneutral reactions due to increasing DADs, but then increase as the reactions become more endergonic due to, however, decreasing DADs (by Hammond's postulate, as discussed above). In other words, a V-shaped
increases from negative through zero to positive values (Fig. 4A). In contrast, ln(kDAD) is expected to decrease as exergonic reactions approach thermoneutral reactions due to increasing DADs, but then increase as the reactions become more endergonic due to, however, decreasing DADs (by Hammond's postulate, as discussed above). In other words, a V-shaped  relationship is expected (Fig. 4B). Since the observed ln(k2)–ΔG° correlation is the “sum” of the two relationships (A and B), the observed ln(k2)–ΔG° correlation is expected to show a broken-line profile with a breaking point at ΔG° = 0 (Fig. 4C).
relationship is expected (Fig. 4B). Since the observed ln(k2)–ΔG° correlation is the “sum” of the two relationships (A and B), the observed ln(k2)–ΔG° correlation is expected to show a broken-line profile with a breaking point at ΔG° = 0 (Fig. 4C).
 | ||
Fig. 4 Predictions from the VA-AHT-inspired model for H-tunneling reactions. (A) The linear  correlation involving both endergonic and exergonic reactions. (B) The V-shaped correlation involving both endergonic and exergonic reactions. (B) The V-shaped  correlation with the turning point at correlation with the turning point at  . (C) The overall ln(k)–ΔG° correlation that reflects the combined assumed equal contributions of the plots (A) and (B); case (a): (A) is a steeper line or (B) is a shallow V-shape or both; from cases (b) to (c): (A) becomes flatter or (B) becomes deeper or both. Note that the Λ-shaped . (C) The overall ln(k)–ΔG° correlation that reflects the combined assumed equal contributions of the plots (A) and (B); case (a): (A) is a steeper line or (B) is a shallow V-shape or both; from cases (b) to (c): (A) becomes flatter or (B) becomes deeper or both. Note that the Λ-shaped  relationship is another prediction from the model, mirroring the V-shaped relationship is another prediction from the model, mirroring the V-shaped  correlation (but kDAD is not directly experimentally accessible). correlation (but kDAD is not directly experimentally accessible). | ||
Theoretically, the pattern of the ln(k2)–ΔG° correlation across exergonic and endergonic reactions depends upon the nature of the reaction systems, specifically, the steepness of the linear  correlation and the depth of the V-shaped
correlation and the depth of the V-shaped  correlation. Fig. 4C(a)–(c) describe three representative patterns. In (a), the
correlation. Fig. 4C(a)–(c) describe three representative patterns. In (a), the  linear correlation is steep while the V-shaped
linear correlation is steep while the V-shaped  correlation is shallow, leading to only a slight deviation at ΔG° = 0. From (b) to (c), the former correlation becomes less steep and/or the latter becomes deeper, leading to a more evident break and, ultimately a distinctly V-shaped profile. Therefore, all three patterns (a) to (c) are possible. Fig. 3A presents the V-shaped ln(k2)–ΔG° correlation (type (c) pattern). This correlation, along with the Λ-shaped ΔEa–ΔG° correlation in Fig. 3B, appears to support the two-coordinate mechanism proposed in the VA-AHT-inspired model.
correlation is shallow, leading to only a slight deviation at ΔG° = 0. From (b) to (c), the former correlation becomes less steep and/or the latter becomes deeper, leading to a more evident break and, ultimately a distinctly V-shaped profile. Therefore, all three patterns (a) to (c) are possible. Fig. 3A presents the V-shaped ln(k2)–ΔG° correlation (type (c) pattern). This correlation, along with the Λ-shaped ΔEa–ΔG° correlation in Fig. 3B, appears to support the two-coordinate mechanism proposed in the VA-AHT-inspired model.
It should be noted that patterns (b) and (c) in Fig. 4C also resemble the Marcus inverted region. It is important, however, to note that relying solely on the ln(k)–ΔG° correlation to identify the DAD sampling mechanism needs caution as the bent or V-shaped  relationship may be masked by the usually dominant and thus much steeper
relationship may be masked by the usually dominant and thus much steeper  relationship. The latter relationship reflects structural rehybridization, charge redistribution, and solvent reorganization (pattern (a)), and thus contribute significantly to the activation process. Furthermore, the “bentness” of the ln(k)–ΔG° correlation depends on the relative linear correlations in between exergonic and endergonic regions. Each slope is influenced by factors such as electronic and steric properties, structural dynamics, the statistical size of the dataset, as well as experimental uncertainties. Consequently, failure to observe a bent or V-shaped ln(k)–ΔG° correlation cannot preclude the existence of a DAD sampling mechanism. Therefore, the seemingly linear ln(k)–ΔG° correlation observed for all of the 34 reactions in Fig. 2A cannot be taken as evidence against the existence of the DAD sampling mechanism. Nevertheless, simultaneous observation of both the Λ-shaped ΔEa–ΔG° relationship and V-shaped ln(k)–ΔG° relationship for the 12 reactions of HEH (Fig. 3B versus 3A) provides strong support for our DAD–ΔEa relationship hypothesis.
relationship. The latter relationship reflects structural rehybridization, charge redistribution, and solvent reorganization (pattern (a)), and thus contribute significantly to the activation process. Furthermore, the “bentness” of the ln(k)–ΔG° correlation depends on the relative linear correlations in between exergonic and endergonic regions. Each slope is influenced by factors such as electronic and steric properties, structural dynamics, the statistical size of the dataset, as well as experimental uncertainties. Consequently, failure to observe a bent or V-shaped ln(k)–ΔG° correlation cannot preclude the existence of a DAD sampling mechanism. Therefore, the seemingly linear ln(k)–ΔG° correlation observed for all of the 34 reactions in Fig. 2A cannot be taken as evidence against the existence of the DAD sampling mechanism. Nevertheless, simultaneous observation of both the Λ-shaped ΔEa–ΔG° relationship and V-shaped ln(k)–ΔG° relationship for the 12 reactions of HEH (Fig. 3B versus 3A) provides strong support for our DAD–ΔEa relationship hypothesis.
Therefore, the Λ-shaped ΔEa–ΔG° correlation and various patterns of the ln(k2)–ΔG° correlation are predicted by the VA-AHT-inspired model, and our experiments are consistent with these predictions. Note that observation of a Λ-shaped ΔEa–ΔG° correlation does not guarantee that an evidently bent or V-shaped ln(k)–ΔG° correlation will also be observed. Likewise, a single bent or V-shaped ln(k2)–ΔG° correlation should also be used cautiously to determine the DAD sampling mechanism.
The Λ-shaped ln(KIE)–ΔG° correlation is also consistent with the Λ-shaped ΔEa–ΔG° relationship
With the KIE data in hand, we are also able to study the KIE–ΔG° correlation. Study of this relationship has had a long history. In 1960, Melander and Westheimer proposed that the maximum KIE should occur at ΔG° = 0 where the transition state (TS) is symmetric.75,76 Later, in 1980, Kresge deduced a parabolic relationship linking KIEs to (ΔG°)2 from the Marcus rate theory.77 In the 1970s, Bell provided an alternative by introducing a tunnel correction to the one-dimensional energy barrier in the TS theory. In his model, tunneling probability is the greatest at a symmetrical TS, leading to the highest KIE, and it decreases as the TS becomes reactant- or product-like.78 Since the 1980s, Kreevoy and coworkers studied the structure–KIE relationship for the hydride transfer reactions of NADH models in isopropanol/water using the Marcus rate theory that attributes a fraction of the KIE to the corner-cutting tunneling mechanism.72,74,79–81 Kil and Lee later found that KIEs of several such exergonic hydride transfer reactions increase as ΔG° becomes less negative close to zero.82 They used Kreevoy's treatment to explain their results and further predicted that the KIE–ΔG° relationship would reverse upon entering the endergonic region, which has, however, not yet been experimentally supported due to lack of experiment data. Thus, this latter model also predicts a similar bell-shaped (or “upward/downward”) dependence of KIEs on ΔG°, with maximal tunneling and thus the largest KIE occurring near ΔG° ∼0, where the effective barrier is highest.Within the VA-AHT(-inspired) model, the KIE is defined differently. It is primarily dependent upon the difference in isotopic wave-function overlaps at the TRSs over a spectrum of DADs.6,8,40,59 Because the vibrational wave-packet of H is more diffuse than that of the D isotope, the H-overlap is more than the D-overlap so that KIE > 140,83 (in other words, because D-tunneling requires shorter DADs than H-tunneling,40,83–86 kD,DAD < kH,DAD). Moreover, since the overlap for D-transfer decreases more rapidly than that for H-transfer with increasing DAD, the KIE increases with DAD (in other words, the difference between kD,DAD and kH,DAD becomes larger as the DAD increases). Consequently, the KIE is predicted to be the largest at ΔG° = 0 where the DAD is the longest, and to decrease as the TRS becomes reactant- or product-like where the DAD shortens. Since the  relationship is V-shaped (Fig. 4B), and kTRS is mostly isotope insensitive, the
relationship is V-shaped (Fig. 4B), and kTRS is mostly isotope insensitive, the  relationship is expected to be Λ-shaped.
relationship is expected to be Λ-shaped.
We examine the ln(KIE)–ΔG° correlation to indirectly test the predicted Λ-shaped  relationship. The 34 ln(KIE)–ΔG° data points and the 12 such data points for the reactions of HEH only, are plotted in Fig. 5A and B, respectively. Linear fits were applied separately to the exergonic and endergonic regions, with the ΔG° = 0 point extrapolated from the exergonic data. In both plots, the endergonic reactions appear to reverse the trend observed for the exergonic reactions. The reverse trend could be further supported by including the smaller KIE values (3.63–4.83) from the endergonic reactions between alcohols and PhXn+ (Rxns 35–38), but again because of lack of the corresponding ΔG° values these KIE data cannot be used for quantitative fitting analysis.
relationship. The 34 ln(KIE)–ΔG° data points and the 12 such data points for the reactions of HEH only, are plotted in Fig. 5A and B, respectively. Linear fits were applied separately to the exergonic and endergonic regions, with the ΔG° = 0 point extrapolated from the exergonic data. In both plots, the endergonic reactions appear to reverse the trend observed for the exergonic reactions. The reverse trend could be further supported by including the smaller KIE values (3.63–4.83) from the endergonic reactions between alcohols and PhXn+ (Rxns 35–38), but again because of lack of the corresponding ΔG° values these KIE data cannot be used for quantitative fitting analysis.
 | ||
| Fig. 5 (A) The linear fit of the ln(KIE(25 °C))–ΔG° data for the 34 hydride transfer reactions. The reactions 35–38 are not included (see the text). (B) The linear fit of the ln(KIE(25 °C))–ΔG° data for the 12 hydride transfer reactions of a common hydride donor HEH (Rxns 23–34). The empty green circle point in each plot is an extrapolated point at ΔG° = 0 derived from the linear equation from the exergonic reactions. | ||
The Λ-shaped ln(KIE)–ΔG° relationship is consistent with the Λ-shaped ΔEa–ΔG° relationship in that both ΔEas and KIEs reach maxima near thermoneutral conditions (Fig. 5 versus 3B). Importantly, both relationships can be uniformly explained using the DAD coordinate mechanism within the VA-AHT-inspired model. According to this model, the KIE increases with the DAD. Note that previous studies on enzymatic reactions as well as NADH/NAD+ model reactions have showcased this KIE–DAD relationship for exergonic reactions (ref. 48 and references cited therein). Therefore, the maxima in both the KIE and ΔEa occurring near ΔG° = 0 correspond to the longest DADs in H-tunneling mechanisms.
We noticed that the slopes of the ΔEa–ΔG° (0.0268) and ln(KIE)–ΔG° (0.0196) correlations for the exergonic reactions are significantly small (Fig. 2B and 5A). This likely partly reflects the fact that ΔEa and ln(KIE) correlate only with the  , which constitutes only a small fraction of the overall ΔG° in this particular class of reactions, so that they are much less sensitive to the overall ΔG°.
, which constitutes only a small fraction of the overall ΔG° in this particular class of reactions, so that they are much less sensitive to the overall ΔG°.
Conclusions
Free-energy dependences of ΔEa, rates (ln(k)), and ln(KIE) were analysed with 34 hydride tunneling reactions of NADH/NAD+ models between two carbons in acetonitrile, which span both exergonic and endergonic regions. A clear linear ΔEa–ΔG° relationship was, for the first time, observed for the exergonic reactions, with ΔEa increasing as ΔG° approaches zero. A quantitative relationship has not yet been obtained for the limited number of endergonic reactions, but reversal of the trend from the exergonic reactions including reactions 35–38 has been clearly shown. This is consistent with our hypothesis regarding the DAD–ΔEa relationship. ΔG° (strictly, ) acts as an indicator of system rigidity (or DAD distributions), where thermoneutral reactions correspond to the most flexible and longest DADs. Overall, a portion of ΔG° of the reactions modulates the CT complexation vibrations and thus DAD sampling, which is directly related to the T-dependence of KIEs. This resulting Λ-shaped ΔEa–ΔG° relationship agrees with the prediction from the VA-AHT-inspired model that involves both the TRS formation and DAD sampling processes and attributes the KIE to only the latter DAD sampling process.
) acts as an indicator of system rigidity (or DAD distributions), where thermoneutral reactions correspond to the most flexible and longest DADs. Overall, a portion of ΔG° of the reactions modulates the CT complexation vibrations and thus DAD sampling, which is directly related to the T-dependence of KIEs. This resulting Λ-shaped ΔEa–ΔG° relationship agrees with the prediction from the VA-AHT-inspired model that involves both the TRS formation and DAD sampling processes and attributes the KIE to only the latter DAD sampling process.
The second prediction from the same model includes the bent- or V-shaped ln(k)–ΔG° relationship with the breaking/turning point at ΔG° = 0. It was found that the ln(k)–ΔG° plot is linear across the 34 reactions, but when using the data from the 12 reactions with a common hydride donor HEH, the V-shaped ln(k)–ΔG° relationship emerges, which mirrors the Λ-shaped ΔEa–ΔG° relationship found from the same series of the reactions. The latter also appears to agree with the DAD sampling mechanism within the VA-AHT-inspired model.
The third prediction from the model includes the Λ-shaped ln(KIE)–ΔG° relationship. It was found that the ln(KIE)–ΔG° plot is linear across the 32 exergonic reactions, but when using the endergonic reactions, the correlation is reversed. This Λ-shaped trend is the same when using the 12 reactions of HEH. These results are consistent with the prediction. Therefore, all of the three predictions from the VA-AHT-inspired model, including the V-shaped ln(k)–ΔG°, Λ-shaped ΔEa–ΔG°, and Λ-shaped ln(KIE)–ΔG° relationships, were simultaneously found in the 12 reactions of HEH, which support our DAD–ΔEa relationship hypothesis.
Caution is required when using the ln(k)–ΔG° correlation alone to identify the DAD sampling mechanism as the expected bent- or V-shaped correlations can be largely masked by the dominant contribution from TRS formation, as well as by system selection and experimental uncertainties. Conversely, observation of the bent- or V-shaped ln(k)–ΔG° correlations alone should also be cautiously used to propose the DAD sampling mechanism, since  could account for a small portion of the ΔG° so that system selection and experimental uncertainty need to be carefully considered. Also, a Λ-shaped ln(KIE)–ΔG° relationship alone should not be considered definitive evidence for DAD sampling either, since similar trends can arise from the Marcus theory combined with the corner-cutting tunneling mechanism process, or even possibly from the classical Melander–Westheimer's “bond-stretching” model. In sharp contrast, the newly identified Λ-shaped ΔEa–ΔG° relationship appearing to arise only from the DAD sampling could provide evidence for the DAD sampling mechanism; however, whether or not this particular relationship is explainable by other tunnelling models remains unclear.
could account for a small portion of the ΔG° so that system selection and experimental uncertainty need to be carefully considered. Also, a Λ-shaped ln(KIE)–ΔG° relationship alone should not be considered definitive evidence for DAD sampling either, since similar trends can arise from the Marcus theory combined with the corner-cutting tunneling mechanism process, or even possibly from the classical Melander–Westheimer's “bond-stretching” model. In sharp contrast, the newly identified Λ-shaped ΔEa–ΔG° relationship appearing to arise only from the DAD sampling could provide evidence for the DAD sampling mechanism; however, whether or not this particular relationship is explainable by other tunnelling models remains unclear.
The small slopes of the Λ-shaped ΔEa–ΔG° and ln(KIE)–ΔG° relationships may suggest that only a small portion of the ΔG° contributes to the activated DAD sampling process. The DAD increases as ΔG° of the exergonic reactions becomes less negative, becomes the largest with thermoneutral reactions, and decreases as ΔG° becomes more positive in the endergonic reactions. This Λ-shaped ΔEa–ΔG° relationship should be useful for evaluating the existing H-tunneling models and helping develop future H-transfer/tunneling theories.
Overall, our ΔEa–ΔG° correlation results provide support for the DAD–ΔEa relationship in both the VA-AHT and VA-AHT-inspired models; systems with more densely distributed small DADs give rise to smaller T-dependence of KIEs. Side-by-side comparison of solution-phase and enzymatic reactions on KIEs and their T-dependences suggests that frequently observed T-independence of KIEs from enzymes can be attributed to strong constructive (fast) protein dynamics (modulated by  ) that compress the donor and acceptor close to each other for H-tunneling to occur. Therefore, there appears a synergy between C–H activation (for TRS formation and DAD sampling) and diverse protein dynamics (encoded in
) that compress the donor and acceptor close to each other for H-tunneling to occur. Therefore, there appears a synergy between C–H activation (for TRS formation and DAD sampling) and diverse protein dynamics (encoded in  and
and , respectively) cooperatively functioning in enzymatic reactions.
, respectively) cooperatively functioning in enzymatic reactions.
Author contributions
A. A.-K., N. D., B. D. and J. S. performed organic synthesis and kinetic measurements. A. A.-K. and N. D. wrote part of the paper. B. D. analyzed the data. L. P. and S. P. performed organic synthesis. Y. L. designed research and wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Data availability
The data underlying this study are available in the published article and its supplementary information (SI). Supplementary information: synthesis, procedures to determine the rate constants, T-dependence of KIE plots, tabulated raw rate constant data, and the primary kinetic experiment data. See DOI: https://doi.org/10.1039/d6sc01847e.Acknowledgements
Acknowledgment is made to the donor of the National Institutes of Health (NIH R15 GM148951). We thank Pratichhya Adhikari (Southern Illinois University Edwardsville) for helping collect part of the kinetic data for the reaction of MPH with p-MeOPhXn+, and Binita Maharajn and Peter Maness (from the same institution) for initial kinetic study of the reaction of MAH with Tr+.References
- A. Kohen, R. Cannio, S. Bartolucci and J. P. Klinman, Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase, Nature, 1999, 399, 496–499 CrossRef CAS PubMed.
- M. J. Knapp, K. Rickert and J. P. Klinman, Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlating hydrogen tunneling with protein dynamics, J. Am. Chem. Soc., 2002, 124, 3865–3874 CrossRef CAS PubMed.
- R. S. Sikorski, L. Wang, K. A. Markham, P. T. R. Rajagopalan, S. J. Benkovic and A. Kohen, Tunneling and coupled motion in the E. coli dihydrofolate reductase catalysis, J. Am. Chem. Soc., 2004, 126, 4778–4779 CrossRef CAS PubMed.
- E. J. Loveridge, R. M. Evans and R. K. Allemann, Solvent Effects on Environmentally Coupled Hydrogen Tunnelling During Catalysis by Dihydrofolate Reductase from Thermotoga maritima, Chem.–Eur. J., 2008, 14(34), 10782–10788 CrossRef CAS PubMed.
- Z. D. Nagel and J. P. Klinman, Update 1 of: Tunneling and dynamics in enzymatic hydride transfer, Chem. Rev., 2010, 110, PR41–PR67 CrossRef PubMed.
- C. R. Pudney, O. L. Johannissen, M. J. Sutcliffe, S. Hay and N. S. Scrutton, Direct Analysis of Donor-Acceptor Distance and Relationship to Isotope Effects and the Force Constant for Barrier Compression in Enzymatic H-Tunneling Reactions, J. Am. Chem. Soc., 2010, 132, 11329–11335 CrossRef CAS.
- Z. Wang, P. N. Singh, M. C. Czekster, A. Kohen and V. L. Schramm, Protein Mass-Modulated Effects in the Catalytic Mechanism of Dihydrofolate Reductase: Beyond Promoting Vibrations, J. Am. Chem. Soc., 2014, 136, 8333–8341 CrossRef CAS PubMed.
- A. Kohen, Role of Dynamics in Enzyme Catalysis: Substantial vs. Semantic Controversies, Acc. Chem. Res., 2015, 48, 466–473 CrossRef CAS PubMed.
- E. Romero, S. T. Ladani, D. Hamelberg and G. Gadda, Solvent-Slaved Motions in the Hydride Tunneling Reaction Catalyzed by Human Glycolate Oxidase, ACS Catal., 2016, 6, 2113–2120 CrossRef CAS.
- J. P. Klinman and A. R. Offenbacher, Understanding Biological Hydrogen Transfer Through the Lens of Temperature Dependent Kinetic Isotope Effects, Acc. Chem. Res., 2018, 51, 1966–1974 CrossRef CAS PubMed.
- G. W. Howe and W. A. van der Donk, Temperature-Independent Kinetic Isotope Effects as Evidence for a Marcus-like Model of Hydride Tunneling in Phosphite Dehydrogenase, Biochemistry, 2019, 58, 4260–4268 CrossRef CAS PubMed.
- P. Singh, A. Vandemeulebroucke, J. Li, C. Schulenburg, G. Fortunato, A. Kohen, D. Hilvert and C. M. Cheatum, Evolution of the Chemical Step in Enzyme Catalysis, ACS Catal., 2021, 11, 6726–6732 CrossRef CAS.
- E. Hatcher, A. V. Soudackov and A. Hammes-Schiffer, Proton-Coupled Electron Transfer in Soybean Lipoxygenase: Dynamical Behavior and Temperature Dependence of Kinetic Isotope Effects, J. Am. Chem. Soc., 2007, 129, 187–196 CrossRef CAS PubMed.
- S. Hu, A. V. Soudackov, S. Hammes-Schiffer and J. P. Klinman, Enhanced Rigidification within a Double Mutant of Soybean Lipoxygenase Provides Experimental Support for Vibronically Nonadiabatic Proton-Coupled Electron Transfer Models, ACS Catal., 2017, 7, 3569–3574 CrossRef CAS.
- S. Hammes-Schiffer and S. J. Benkovic, Relating protein motion to catalysis, Annu. Rev. Biochem., 2006, 75, 519–541 CrossRef CAS PubMed.
- K. A. Henzler-Wildman, M. Lei, V. Thai, S. J. Kerns, M. Karplus and D. Kern, A Hierarchy of Timescales in Protein Dynamics Is Linked to Enzyme Catalysis, Nature, 2007, 450, 913–916 CrossRef CAS PubMed.
- S. Hay and N. S. Scruton, Good vibrations in enzyme-catalysed reactions, Nat. Chem., 2012, 4, 161–168 CrossRef CAS.
- J. P. Klinman and A. Kohen, Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis, Annu. Rev. Biochem., 2013, 82, 471–496 CrossRef CAS PubMed.
- V. L. Schramm and S. D. Schwartz, Promoting vibrations and the function of enzymes. Emerging theoretical and experimental convergence, Biochemistry, 2018, 57, 3299–3308 CrossRef CAS PubMed.
- J. P. Klinman, S. M. Miller and N. G. J. Richards, A Foundational Shift in Models for Enzyme Function, J. Am. Chem. Soc., 2025, 147, 14884–14904 CrossRef CAS PubMed.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Promoting motions in enzyme catalysis probed by pressure studies of kinetic isotope effects, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 507–512 CrossRef CAS.
- J. Bandaria, S. Dutta, M. W. Nydegger, W. Rock, A. Kohen and C. Cheatum, Characterizing the dynamics of functionally relevant complexes of formate dehydrogenase, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17974–17979 CrossRef CAS PubMed.
- E. J. Loveridge, L.-H. Tey and R. K. Allemann, Solvent Effects on Catalysis by Escherichia coli Dihydrofolate Reductase, J. Am. Chem. Soc., 2010, 132, 1137–1143 CrossRef CAS PubMed.
- Q. Liu, Y. Zhao, B. Hammann, J. Eilers, Y. Lu and A. Kohen, A Model Reaction Assesses Contribution of H-Tunneling and Coupled Motions to Enzyme Catalysis, J. Org. Chem., 2012, 77, 6825–6833 CrossRef CAS PubMed.
- Y. Lu, S. Wilhelm, M. Bai, P. Maness and L. Ma, Replication of the Enzymatic Temperature Dependency of the Primary Hydride Kinetic Isotope Effects in Solution: Caused by the Protein Controlled Rigidity of the Donor-Acceptor Centers?, Biochemistry, 2019, 58, 4035–4046 CrossRef CAS PubMed.
- D. Antoniou and S. D. Schwartz, Role of protein motions in catalysis by Formate Dehydrogenase, J. Phys. Chem. B, 2020, 124, 9483–9489 CrossRef CAS PubMed.
- M. Bai, P. Rijal, S. Salarvand and Y. Lu, Correlation of Temperature Dependence of Hydride Kinetic Isotope Effects with Donor-Acceptor Distances in Two Solvents of Different Polarities, Org. Biomol. Chem., 2023, 21, 5090–5097 RSC.
- S. D. Schwartz, Protein Dynamics and Enzymatic Catalysis, J. Phys. Chem. B, 2023, 127, 2649–2660 CrossRef CAS PubMed.
- G. Singh, A. Austin, M. Bai, J. Bradshaw, B. A. Hammann, D. E. K. Kabotso and Y. Lu, Study of the Effects of Remote Heavy Group Vibrations on the Temperature Dependence of Hydride Kinetic Isotope Effects of the NADH/NAD+ Model Reactions, ACS Omega, 2024, 9, 20593–20600 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Explaining Kinetic Isotope Effects in Proton-Coupled Electron Transfer Reactions, Acc. Chem. Res., 2025, 58, 1335–1344 CrossRef CAS PubMed.
- S. Hu, A. R. Offenbacher, E. M. Thompson, C. L. Gee, J. Wilcoxen, C. A. M. Carr, D. M. Prigozhin, V. Yang, T. Alber, R. D. Britt, J. S. Fraser and J. P. Klinman, Biophysical Characterization of a Disabled Double Mutant of Soybean Lipoxygenase: The “Undoing” of Precise Substrate Positioning Relative to Metal Cofactor and an Identified Dynamical Network, J. Am. Chem. Soc., 2019, 141, 1555–1567 CrossRef CAS PubMed.
- J. Pu, S. Ma, J. Gao and D. G. Truhlar, Small temperature dependence of the kinetic isotope effect for the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase, J. Phys. Chem. B, 2005, 109, 8551–8556 CrossRef CAS PubMed.
- H. Liu and A. Warshel, Origin of the Temperature Dependence of Isotope Effects in Enzymatic Reactions: The Case of Dihydrofolate Reductase, J. Phys. Chem. B, 2007, 111, 7852–7861 CrossRef CAS PubMed.
- S. C. L. Kamerlin and A. Warshel, An analysis of all the relevant facts and arguments indicates that enzyme catalysis does not involve large contributions from nuclear tunneling, J. Phys. Org. Chem., 2010, 23, 677–684 CrossRef CAS PubMed.
- R. A. More O'Ferrall, Introduction to a symposium in print on tunnelling, J. Phys. Org. Chem., 2010, 23, 559–560 CrossRef.
- G. D. Truhlar, Tunneling in enzymatic and nonenzymatic hydrogen transfer reactions, J. Phys. Org. Chem., 2010, 23, 660–676 CrossRef.
- Z. Wang and A. Kohen, Thymidylate synthase catalyzed H-transfers: Two chapters in one tale, J. Am. Chem. Soc., 2010, 132, 9820–9825 CrossRef CAS PubMed.
- C. Ranasinghe, P. Pagano, P. J. Sapienza, A. L. Lee, A. Kohen and C. M. Cheatum, Isotopic Labeling of Formate Dehydrogenase Perturbs the Protein Dynamics, J. Phys. Chem. B, 2019, 123, 10403–10409 CrossRef CAS PubMed.
- M. Horitani, A. R. Offenbacher, C. A. Marcus Carr, T. Yu, V. Hoeke, G. E. Cutsail, S. Hammes-Schiffer, J. P. Klinman and B. M. Hoffman, 13C ENDOR Spectroscopy of Lipoxygenase−Substrate Complexes Reveals the Structural Basis for C−H Activation by Tunneling, J. Am. Chem. Soc., 2017, 139, 1984–1997 CrossRef CAS PubMed.
- D. Roston and A. Kohen, Elusive transition state of alcohol dehydrogenase unveiled, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9572–9577 CrossRef CAS PubMed.
- N. Kanaan, S. Ferrer, S. Martí, M. Garcia-Viloca, A. Kohen and V. Moliner, Temperature Dependence of the Kinetic Isotope Effects in Thymidylate Synthase. A Theoretical Study, J. Am. Chem. Soc., 2011, 133, 6692–6702 CrossRef CAS PubMed.
- A. R. Mhashal and D. T. Major, Temperature-Dependent Kinetic Isotope Effects in R67 Dihydrofolate Reductase from Path-Integral Simulations, J. Phys. Chem. B, 2021, 125, 1369–1377 CrossRef CAS PubMed.
- J. P. Layfield and S. Hammes-Schiffer, Hydrogen tunneling in enzymes and biomimetic models, Chem. Rev., 2014, 114, 3466–3494 CrossRef CAS PubMed.
- P. Maness, S. Koirala, P. Adhikari, N. Salimraftar and Y. Lu, Substituent Effects on Temperature Dependence of Kinetic Isotope Effects in Hydride-Transfer Reactions of NADH/NAD+ Analogues in Solution: Reaction Center Rigidity Is the Key, Org. Lett., 2020, 22, 5963–5967 CrossRef CAS PubMed.
- M. Bai, S. Koirala and Y. Lu, Direct Correlation Between Donor-Acceptor Distance and Temperature Dependence of Kinetic Isotope Effects in Hydride-Tunneling Reactions of NADH/NAD+ Analogues, J. Org. Chem., 2021, 86, 7500–7507 CrossRef CAS PubMed.
- P. Adhikari, M. Song, M. Bai, P. Rijal, N. DeGroot and Y. Lu, Solvent Effects on the Temperature Dependence of Hydride Kinetic Isotope Effects: Correlation to the Donor–Acceptor Distances, J. Phys. Chem. A, 2022, 126, 7675–7686 CrossRef CAS PubMed.
- A. Beach, P. Adhikari, G. Singh, M. Song, N. DeGroot and Y. Lu, Structural Effects on the Temperature Dependence of Hydride Kinetic Isotope Effects of the NADH/NAD+ Model Reactions in Acetonitrile: Charge-Transfer Complex Tightness Is a Key, J. Org. Chem., 2024, 89, 3184–3193 CrossRef CAS PubMed.
- A. Austin, J. Sager, L. Phan and Y. Lu, Structural Effects on the Hydride-Tunneling Kinetic Isotope Effects of NADH/NAD+ Model Reactions: Relating to the Donor–Acceptor Distances, J. Org. Chem., 2025, 90, 3110–3115 CrossRef CAS PubMed.
- M. Bai, G. Singh and Y. Lu, Rigidity Analysis of Hydride Tunneling Ready States from Secondary Kinetic Isotope Effects and Hammett Correlations: Relating to the Temperature Dependence of Kinetic Isotope Effects, J. Phys. Org. Chem., 2025, 38 DOI:10.1002/poc.70002.
- B. Pokhrel, J. Sager, P. Adhikari, G. Singh, B. Dhakal and Y. Lu, Large Temperature Dependence of Large Kinetic Isotope Effects of Multistep Hydride Reduction of p-Chloranil by NADH Models in Acetonitrile: Proton Tunneling within Loose Radical Ion-Pairs, J. Org. Chem., 2025, 16912–16917 CrossRef CAS PubMed.
- D. Roston, C. M. Cheatum and A. Kohen, Hydrogen Donor-Acceptor Fluctuations from Kinetic Isotope Effects: A Phenomenological Model, Biochemistry, 2012, 51, 6860–6870 CrossRef CAS PubMed.
- V. Stojkoviç, L. Perissinotti, D. Willmer, S. Benkovic and A. Kohen, Effects of the donor acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction, J. Am. Chem. Soc., 2012, 134, 1738–1745 CrossRef PubMed.
- P. Pagano, Q. Guo, C. Ranasinghe, E. Schroeder, K. Robben, F. Häse, H. Ye, K. Wickersham, A. Aspuru-Guzik, D. T. Major, L. Gakhar, A. Kohen and C. M. Cheatum, Oscillatory Active-Site Motions Correlate with Kinetic Isotope Effects in Formate Dehydrogenase, ACS Catal., 2019, 9, 11199–11206 CrossRef CAS PubMed.
- M. J. Knapp and J. P. Klinman, Environmentally coupled hydrogen tunneling. Linking catalysis to dynamics, Eur. J. Biochem., 2002, 269, 3113–3121 CrossRef CAS PubMed.
- N. Agrawal, B. Hong, C. Mihai and A. Kohen, Vibrationally Enhanced Hydrogen Tunneling in the Escherichia coli Thymidylate Synthase Catalyzed Reaction, Biochemistry, 2004, 43, 1998–2006 CrossRef CAS PubMed.
- Z. D. Nagel, C. W. Meadows, M. Dong, B. J. Bahnson and J. P. Klinman, Active Site Hydrophobic Residues Impact Hydrogen Tunneling Differently in a Thermophilic Alcohol Dehydrogenase at Optimal versus Nonoptimal Temperatures, Biochemistry, 2012, 51, 4147–4156 CrossRef CAS PubMed.
- K. Francis, P. Sapienza, A. Lee and A. Kohen, The Effect of Protein Mass Modulation on Human Dihydrofolate Reductase, Biochemistry, 2016, 55, 1100–1106 CrossRef CAS PubMed.
- A. Geddes, C. E. Paul, S. Hay, F. Hollmann and N. S. Scrutton, Donor−Acceptor Distance Sampling Enhances the Performance of “Better than Nature” Nicotinamide Coenzyme Biomimetics, J. Am. Chem. Soc., 2016, 138, 11089–11092 CrossRef CAS PubMed.
- J. P. Klinman, A new model for the origin of kinetic hydrogen isotope effects, J. Phys. Org. Chem., 2010, 23, 606–612 CrossRef CAS.
- R. K. Roy, D. Antoniou and S. D. Schwartz, The Shaping of Enzymatic Free Energy Barriers through the Creation of Rate-Promoting Vibrations via Inter-Residue Cross-Talk on Multiple Time Scales, J. Phys. Chem. B, 2025, 129, 9973–9982 CrossRef CAS PubMed.
- M. Delgado, S. Görlich, J. E. Longbotham, N. S. Scrutton, S. Hay, V. Moliner and I. Tuñón, Convergence of Theory and Experiment on the Role of Preorganization, Quantum Tunneling, and Enzyme Motions into Flavoenzyme-Catalyzed Hydride Transfer, ACS Catal., 2017, 7, 3190–3198 CrossRef CAS PubMed.
- P. K. Agarwal, S. R. Billeter, P. T. R. Rajagopalan, S. J. Benkovic and S. Hammes-Schiffer, Network of coupled promoting motions in enzyme catalysis, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2794–2799 CrossRef CAS PubMed.
- E. Hatcher, A. V. Soudackov and S. Hammes-Schiffer, Proton-coupled electron transfer in soybean lipoxygenase, J. Am. Chem. Soc., 2004, 126, 5763–5775 CrossRef CAS PubMed.
- B. Salna, A. Benabbas, D. Russo and P. M. Champion, Tunneling kinetics and nonadiabatic proton-coupled electron transfer in proteins: The effect of electric fields and anharmonic donor-acceptor interactions, J. Phys. Chem. B, 2017, 121, 6869–6881 CrossRef CAS PubMed.
- P. Li, A. V. Soudackov and S. Hammes-Schiffer, Fundamental insights into proton-coupled electron transfer in soybean lipoxygenase from quantum mechanical/molecular mechanics free energy simulations, J. Am. Chem. Soc., 2018, 140, 3068–3076 CrossRef CAS PubMed.
- J. Pang, J. Pu, J. Gao, D. G. Truhlar and R. K. Allemann, Hydride Transfer Reaction Catalyzed by Hyperthermophilic Dihydrofolate Reductase Is Dominated by Quantum Mechanical Tunneling and Is Promoted by Both Inter- and Intramonomeric Correlated Motions, J. Am. Chem. Soc., 2006, 128, 8015–8023 CrossRef CAS PubMed.
- X.-Q. Zhu, H.-L. Zou, P.-W. Yuan, Y. Liu, L. Cao and J.-P. Cheng, A detailed investigation into the oxidation mechanism of Hantzsch 1,4-dihydropyridines by ethyl -cyanocinnamates and benzylidenemalononitriles, J. Chem. Soc., Perkin Trans. 2, 2000, 1857–1861 RSC.
- G. B. Shen, K. Xia, X. T. Li, J. L. Li, Y. H. Fu, L. Yuan and X. Q. Zhu, Prediction of Kinetic Isotope Effects for Various Hydride Transfer Reactions Using a New Kinetic Model, J. Phys. Chem. A, 2016, 120, 1779–1799 CrossRef CAS PubMed.
- Y. Lu, F. Qu, B. Moore, D. Endicott and W. Kuester, Hydride Reduction of NAD+ Analogues by Isopropyl Alcohol: Kinetics, Deuterium Isotope Effects and Mechanism, J. Org. Chem., 2008, 73, 4763–4770 CrossRef CAS PubMed.
- Y. Lu, F. Qu, Y. Zhao, A. M. Small, J. Bradshaw and B. Moore, Kinetics of the hydride reduction of an NAD(+) analogue by isopropyl alcohol in aqueous and acetonitrile solutions: solvent effects, deuterium isotope effects, and mechanism, J. Org. Chem., 2009, 74, 6503–6510 CrossRef CAS PubMed.
- Y. Lu, J. Bradshaw, Y. Zhao, W. Kuester and D. Kabotso, Structure–reactivity relationship for alcohol oxidations via hydride transfer to a carbocationic oxidizing agent, J. Phys. Org. Chem., 2011, 24, 1172–1178 CrossRef CAS.
- M. M. Kreevoy, D. Ostovic and D. G. Truhlar, Phenomenological manifestations of large-curvature tunneling in hydride-transfer reactions, J. Phys. Chem., 1986, 90, 3766–3774 CrossRef CAS.
- A. Kohen and J. P. Klinman, Enzyme catalysis: beyond classical paradigms, Acc. Chem. Res., 1998, 31, 397–404 CrossRef CAS.
- I.-S. Han Lee, E. H. Jeoung and M. M. Kreevoy, Primary Kinetic Isotope Effects on Hydride Transfer from 1,3-Dimethyl-2-phenylbenzimidazoline to NAD+ Analogues, J. Am. Chem. Soc., 2001, 123, 7492–7496 CrossRef PubMed.
- L. Melander, in Isotope Effects on Reaction Rates, Ronald Press, New York, 1960, pp. 24–32 Search PubMed.
- F. H. Westheimer, THE mangitude of the Primary kinetic isotope effect for compounds of hydrogen and deuterium, Chem. Rev., 1961, 61, 265–273 CrossRef CAS.
- M. M. Kreevoy and S. W. Oh, Relation between rate and equilibrium constants for proton-transfer reactions, J. Am. Chem. Soc., 1973, 95, 4805–4810 CrossRef CAS.
- R. P. Bell, Liversidge lecture. Recent advances in the study of kinetic hydrogen isotope effects, Chem. Soc. Rev., 1974, 3, 513–544 RSC.
- Y. Kim, D. G. Truhlar and M. M. Kreevoy, An experimentally based family of potential energy surfaces for hydride transfer between NAD+ analogs, J. Am. Chem. Soc., 1991, 113, 7837–7847 CrossRef CAS.
- Y. Kim and M. M. Kreevoy, The experimental manifestations of corner-cutting tunneling, J. Am. Chem. Soc., 1992, 114, 7116–7123 CrossRef CAS.
- I.-S. H. Lee, E. H. Jeoung and M. M. Kreevoy, Marcus Theory of a Parallel Effect on R for Hydride Transfer Reaction between NAD+ Analogues, J. Am. Chem. Soc., 1997, 119, 2722–2728 CrossRef CAS.
- H. J. Kil and I.-S. H. Lee, Primary Kinetic Isotope Effects on Hydride Transfer from Heterocyclic Compounds to NAD + Analogues, J. Phys. Chem. A, 2009, 113, 10704–10709 CrossRef CAS PubMed.
- D. Roston and A. Kohen, A Critical Test of the “Tunneling and Coupled Motion” Concept in Enzymatic Alcohol Oxidation, J. Am. Chem. Soc., 2013, 135, 13624–13627 CrossRef CAS PubMed.
- S. Kashefolgheta, M. Razzaghi, B. Hammann, J. Eilers, D. Roston and Y. Lu, Computational Replication of the Abnormal Secondary Kinetic Isotope Effects in a Hydride Transfer Reaction in Solution with a Motion Assisted H-Tunneling Model, J. Org. Chem., 2014, 79, 1989–1994 CrossRef CAS PubMed.
- B. Maharjan, M. R. Boroujeni, J. Lefton, O. R. White, M. Razzaghi, B. A. Hammann, M. Derakhshani-Molayousefi, J. E. Eilers and Y. Lu, Steric Effects on the Primary Isotope Dependence of Secondary Kinetic Isotope Effects in Hydride Transfer Reactions in Solution: Caused by the Isotopically Different Tunneling Ready State Conformations?, J. Am. Chem. Soc., 2015, 137, 6653–6661 CrossRef CAS PubMed.
- M. Derakhshani-Molayousefi, S. Kashefolgheta, J. E. Eilers and Y. Lu, Computational Replication of the Primary Isotope Dependence of Secondary Kinetic Isotope Effects in Solution Hydride-Transfer Reactions: Supporting the Isotopically Different Tunneling Ready State Conformations, J. Phys. Chem. A, 2016, 120, 4277–4284 CrossRef CAS PubMed.
Footnote |
| † A. A. and N. D. contributed equally to both the experiments and writing of the manuscript. |
| This journal is © The Royal Society of Chemistry 2026 |