DOI:
10.1039/D6SC00612D
(Edge Article)
Chem. Sci., 2026,
17, 7082-7090
Atomic Ce sites promote a four-electron pathway of Pt as NADH oxidase mimics for in situ coenzyme regeneration
Received
22nd January 2026
, Accepted 4th February 2026
First published on 16th February 2026
Abstract
Nicotinamide adenine dinucleotide (NAD+) is a key coenzyme for human redox reactions, vital for cellular health and metabolic balance. Lots of NAD+-dependent redox enzymes, like alcohol dehydrogenase (ADH) and lactate dehydrogenase, can catalyze both forward and reverse reactions. However, substrate accumulation and inadequate NAD+ replenishment hinder forward reactions, disrupting the proper metabolism of these substrates. In this work, we reported atomic Ce-doped Pt (Ce1Pt) nanoparticles with abundant oxygen vacancy sites to boost NADH oxidase (NOX)-like activity for coenzyme regeneration. Mechanistic studies reveal that atomic Ce doping increases electron density of Pt and surface defects, enhancing O2 adsorption and accelerating the rate-limiting step. Furthermore, Ce1Pt employs a 4e− pathway for O2 reduction during NADH oxidation, minimizing toxic H2O2 byproducts and improving detection accuracy by reducing oxidative interference. Finally, Ce1Pt enables NAD+ regeneration and substrate metabolism, offering a promising strategy to counteract excessive alcohol intake or lactate accumulation. Through competitive adsorption between the reduced coenzyme NADH of ADH and chromogenic substrates, a microfluidic device integrated with immobilized Ce1Pt achieves blood alcohol detection with a low limit of detection of 0.012 mM.
Introduction
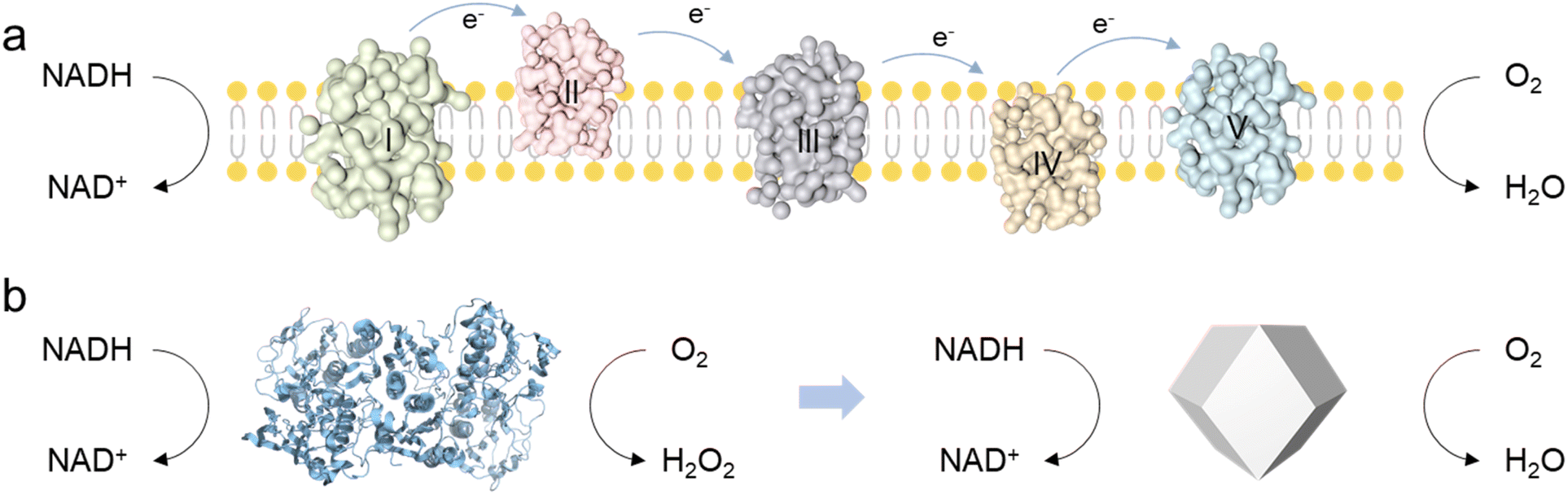
Nicotinamide adenine dinucleotide (NAD+) plays a crucial role in the human body by participating in various biochemical reactions, particularly in cellular metabolism and energy production.1 The human body regulates energy metabolism, redox reactions, and overall cellular health by maintaining the balance between NAD+ and NADH.2,3 However, impairment of the body's health, for instance, due to excessive alcohol intake or lactic acid accumulation, can lead to a considerable increase in NADH levels, disrupting the balance of the NAD+/NADH ratio.4–8 In the human body, the biological functions of NOX are typically performed by other enzymes within a complex electron transport chain, which requires the coordinated action of multiple substrates and cannot catalyze NADH oxidation independently.9 In contrast, NOX from fungi or plants possess a much simpler catalytic architecture, enabling more direct NADH oxidation (Scheme 1).10 However, these NOX can reduce O2 to produce H2O2 for their physiological roles, which do not fulfill the requirements for efficient catalysis and byproduct-free reactions essential for human biological processes. To address this issue, researchers are eagerly exploring NOX mimics as potential alternatives.
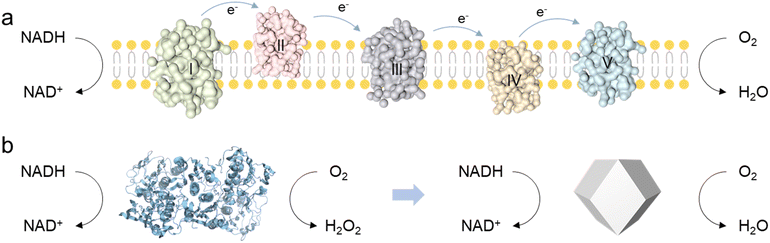 |
| | Scheme 1 (a) The NADH oxidative respiratory chain in the human body. (b) The catalytic pathway of NOX extracted from fungi or plants, and the proposed Ce1Pt for NADH oxidation. | |
Nanozymes as oxidase mimics have been well developed, which can activate O2 and transfer electrons from substrates to O2.11–16 NOX-driven catalytic reactions contain two individual half-reactions: NADH dehydrogenation and O2 reduction (2e− or 4e− pathways).17,18 The designed nanozyme should not only serve as an electron carrier, accepting electrons from NADH and transferring them to O2, but also enable the catalysis of the more efficient 4e− oxygen reduction pathway.19 Due to tunable electronic structures and versatile active sites, noble metal nanomaterials exhibit superior enzyme-like activity and exceptional stability under harsh conditions.20,21 However, research into noble metal nanomaterials as NADH oxidase mimics and their catalytic mechanisms remains largely unexplored.22 This limitation stems from the considerable challenge of constructing electron transport structures (like Fe–S clusters or Cu ions) like natural NOX enzymes. As a result, their ability to activate both NADH and O2 is constrained and the efficiency of proton and electron transfer between NADH and O2 is significantly compromised.10,22 More importantly, their limited electron transfer and utilization efficiency in redox reactions promote the generation of H2O2, which not only interferes with sensing accuracy but also limits their future safety in biological applications.
Herein, we synthesized atomic Ce-doped Ce1Pt with abundant oxygen vacancy (OV) sites to boost the NOX-like activity for coenzyme regeneration. Experimental investigations demonstrated that the resultant Ce1Pt possesses superior NOX-like activity to pristine Pt. Mechanism investigation indicates that the atomically dispersed Ce enhances the electron density of Pt and facilitates the formation of OV sites, thereby effectively promoting substrate adsorption. Unlike the 2e− pathway seen in natural NOX, Ce1Pt catalyzes the oxidation of NADH while facilitating the 4e− reduction of oxygen, significantly enhancing the utilization of electrons extracted from NADH. Through synergistic integration with NAD+-dependent natural enzyme systems, the as-engineered Ce1Pt nanozyme demonstrated efficient cofactor recycling, enabling continuous bioconversion of metabolic substrates, including ethanol, acetaldehyde, and lactate. Then, a microfluidic platform was constructed for the monitoring of blood alcohol with a low limit of detection of 0.012 mM.
Results and discussion
Synthesis and morphology characterization of nanozymes
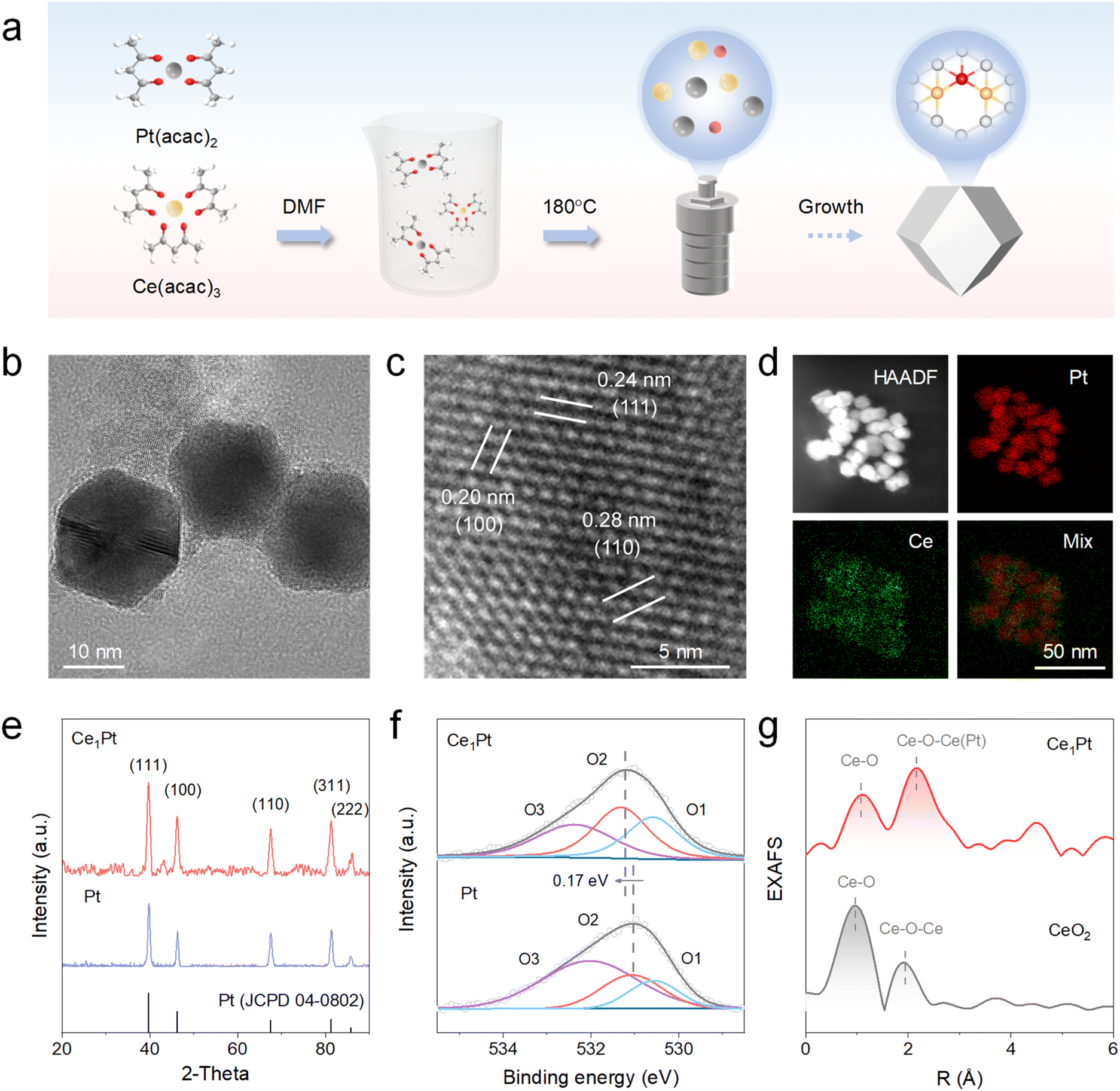
By using platinum(II) acetylacetonate (Pt(acac)2) and cerium(III) acetylacetonate (Ce(acac)3) as precursors, Ce1Pt was successfully synthesized by a one-step hydrothermal method at 180 °C for 12 h (Fig. 1a).23 As a control, Pt was synthesized using the same method, but only Pt(acac)2 was used as the precursor. The morphology of the resultant Ce1Pt, which possesses a uniform cuboctahedron morphology with an average size of ∼20 nm, was characterized by transmission electron microscopy (TEM) (Fig. 1b). For comparison, Pt exhibits cubic morphology with a size similar to that of Ce1Pt. The morphological changes imply that the introduction of Ce enhances the epitaxial growth of the crystal plane, leading to a transformation into the crystal phase structure of Pt (Fig. S1). As shown in Fig. 1c and S2b, high-resolution TEM (HRTEM) images reveal distinct lattice fringes of Ce1Pt, with interplanar spacings of 0.24 nm, 0.20 nm, and 0.28 nm, corresponding to the (111), (100), and (110) planes of Pt. In addition, energy-dispersive X-ray spectroscopy (EDS) analysis verifies the uniform distribution of Ce and Pt within the Ce1Pt nanoparticles (Fig. 1d). The crystal structure of Ce1Pt was thoroughly characterized using X-ray diffraction (XRD) (Fig. 1e). Five diffraction peaks observed at 39.8°, 46.2°, 67.5°, 81.3°, and 85.7° correspond to the (111), (100), (110), (311), and (222) planes of Pt (PDF#04-0802), indicating the consistent crystal structure of Pt. Further analysis using X-ray photoelectron spectroscopy (XPS) revealed that the high-resolution Pt 4f orbital can be divided into two paired peaks corresponding to Pt0 and Pt2+, indicating that platinum predominantly exists in its metallic state in both nanozymes (Fig. S3).24 The Pt 4f orbital of Ce1Pt exhibits a shift of 0.15 eV towards lower binding energy in comparison to the Pt nanozyme, which indicates that electrons are transferred from Ce atoms to Pt atoms and Pt atoms are enriched with electrons. As shown in Figure1f, three characteristic XPS peaks at approximately 530.6 eV, 531.3 eV, and 532.4 eV correspond to lattice oxygen, oxygen defects, and surface-adsorbed oxygen species, respectively.25 The content of oxygen defects in Ce1Pt is significantly higher than that in Pt, indicating that the introduction of Ce effectively removes certain oxygen elements, leading to the formation of more OVs within the Pt lattice.26–28 Besides, electron paramagnetic resonance (EPR) displays a prominent Lorentzian line at a g value of around 2.003, suggesting a high density of unpaired electrons and defects in Ce1Pt (Fig. S4).29,30 As shown in Fig. 1g, the absence of significant Ce–Ce signals in the Fourier transform extended X-ray absorption fine structure (EXAFS) analysis further confirms the atomic dispersion of Ce atoms in Ce1Pt (Fig. 1g).
 |
| | Fig. 1 (a) Schematic illustration for the synthesis of Ce1Pt. (b) TEM image, (c) HRTEM image, (d) HAADF-STEM image, and the corresponding EDS mapping images of Ce1Pt. (e) XRD patterns, (f) O 2p XPS spectra of nanozymes, and (g) EXAFS spectra of Ce1Pt and CeO2. | |
NADH oxidase-like activity of nanozymes
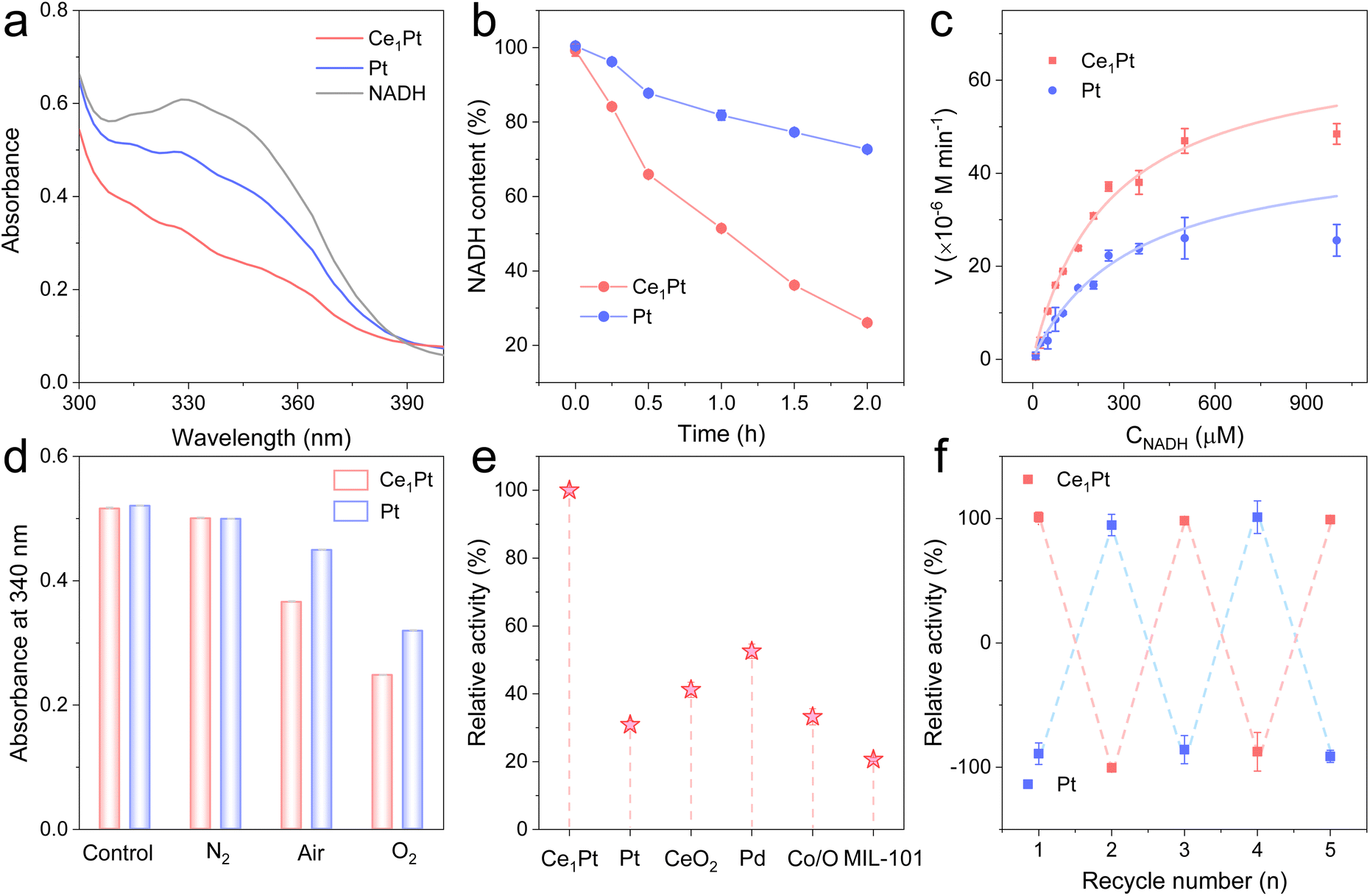
Then, we systematically investigated the NOX-like activity of the resultant nanozymes. NADH, serving as a substrate for NOX, displays a characteristic UV absorption peak at 340 nm. Upon oxidation, the absorbance at this wavelength decreases, enabling the evaluation of the NOX-like activity of the nanozymes. As shown in Fig. 2a and b, Ce1Pt can significantly lower the characteristic peak of NADH at 340 nm in comparison to Pt, indicating that the incorporation of Ce enhances the NOX-like activity of nanozyme.31–33 The NOX-like activity of Ce1Pt exhibits a volcano-shaped dependence on pH, reaching maximum efficiency at pH 7.4, which aligns with physiological conditions (Fig. S5). Under the optimized pH conditions, kinetic experiments indicate that the nanozyme-involved catalytic reaction follows the Michaelis–Menten equation (Fig. 2c). The maximum reaction rate (Vmax) of Ce1Pt is 7.02 × 10−5 M min−1, which is 1.24 times higher than that of Pt. Moreover, Ce1Pt has a lower Michaelis constant (Km), indicating a stronger affinity for the catalytic substrate in comparison to Pt (Table S1).34,35 Notably, the kinetic parameters of Ce1Pt surpass those reported for the majority of previously documented nanozymes. In O2-saturated solutions, the oxidation rate of NADH is higher than that in N2 and air-saturated solutions, confirming the essential role of O2 in the simulated oxidase reaction (Fig. 2d). To evaluate the advantages of Ce1Pt, we compared it with several common nanozymes known for their NOX-like activity. The results demonstrated that it shows a significant advantage in NOX-like activity over other precious metals, metal oxides, and metal–organic frameworks (MOFs) (Fig. 2e). Moreover, Ce1Pt benefits from the inherent stability of noble metals, allowing them to retain catalytic performance after being soaked or dried for 7 days. They are highly resistant to acidic and basic conditions and can be stored at room temperature for long periods without loss of activity (Fig. S6). Even after five cycles, Ce1Pt and Pt retain their catalytic activity, highlighting their high stability (Fig. 2f).
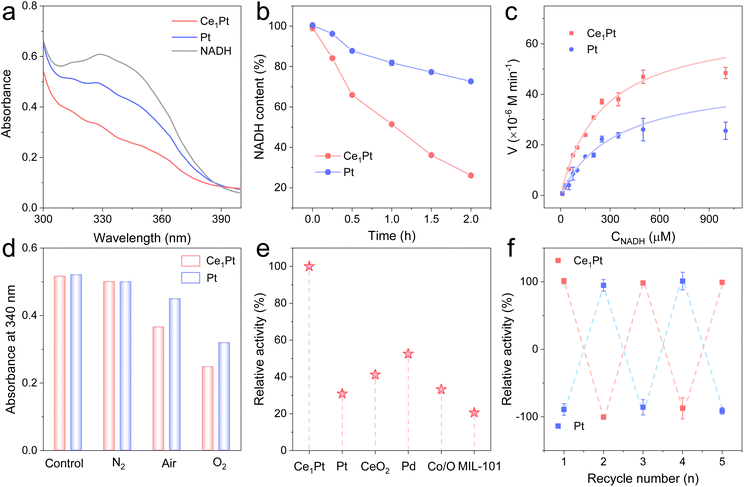 |
| | Fig. 2 (a) Absorption spectra and (b) time-dependent absorbance changes of the NADH oxidation reaction catalyzed by nanozymes. (c) The kinetic curve of nanozymes toward NADH. (d) UV-vis absorption at 340 nm of Ce1Pt-catalyzed NADH oxidation in the N2-, air-, and O2-saturated solutions. (e) The relative NOX-like activity of different nanozymes. (f) The cycling stability of nanozymes. | |
Insights into the underlying catalytic mechanism
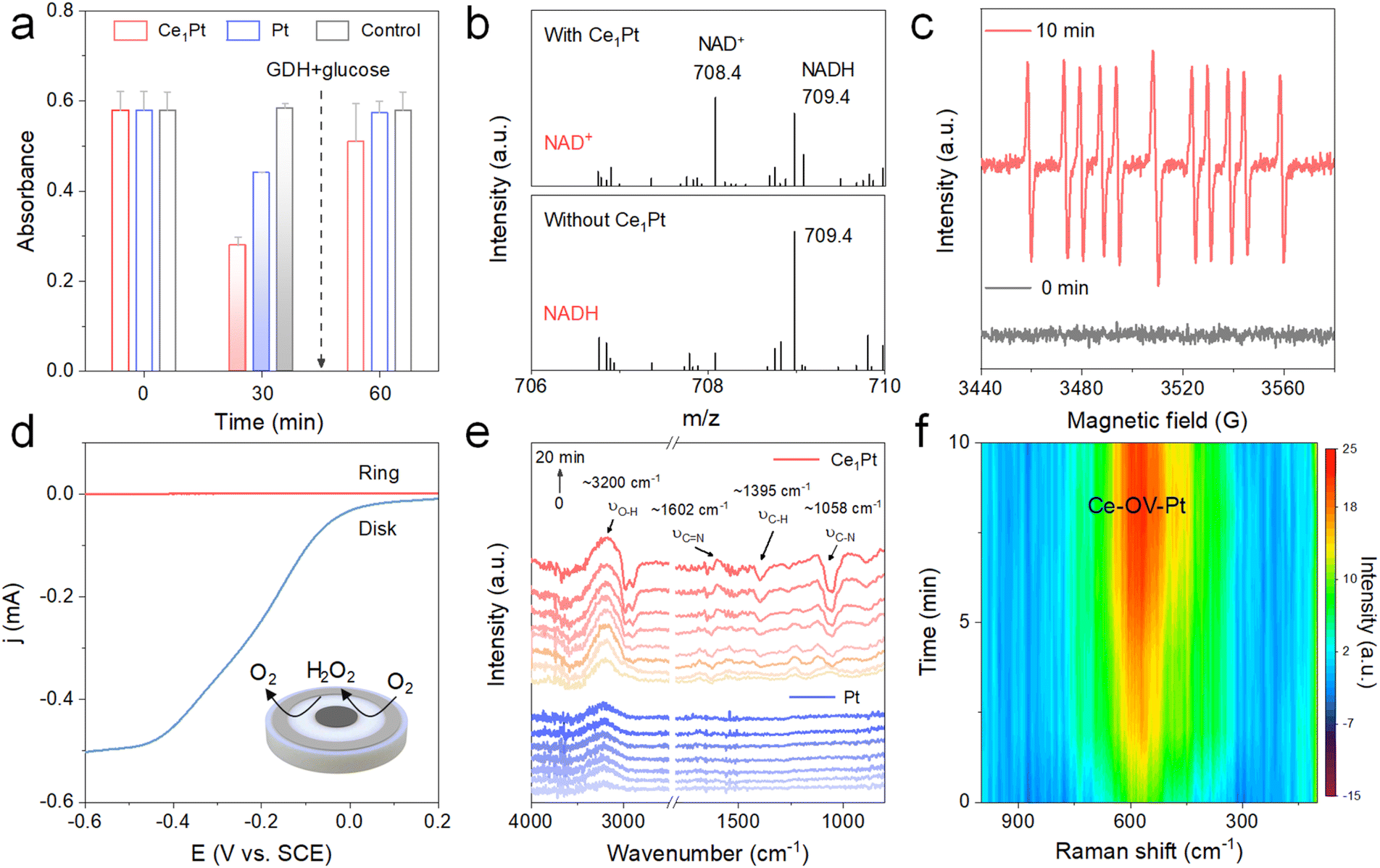
To verify the biological activity of regenerated coenzyme, we introduced glucose and glucose dehydrogenase (GDH) into the reaction system (Fig. 3a and S7). It is observed that after a 30-minute reaction between NADH and the nanozymes, the absorbance at 340 nm decreases, indicating that both Ce1Pt and Pt can effectively oxidize NADH to NAD+ during the process. Then, after the removal of the nanozymes, the addition of glucose and GDH leads to a renewed increase in absorbance. As shown in Fig. 3b, mass spectrometry analysis shows the changes in molecular mass in the solution before and after the reaction. Before the reaction, the mass spectrum displayed only a peak at 709.4, corresponding to the molecular mass of NADH. After the reaction, the peak at 709.4 significantly weakened, and a new peak appeared at 708.4, corresponding to the molecular mass of NAD+, which indicates that Ce1Pt can oxidize NADH to NAD+. Using 5-(2-Carboxyphenyl)-5-methyl-1-pyrroline-N-oxide (CYPMPO) as a spin-trapping agent, EPR was employed to detect the free radicals generated during the Ce1Pt-catalyzed oxidation of NADH. As shown in Fig. 3c, a strong signal of the CYPMPO-NAD adduct was detected by EPR after 10 minutes of reaction catalyzed by Ce1Pt, confirming the formation of the NAD˙ intermediate. Then, electrochemical measurement was used to examine the selectivity of O2 reduction on nanozymes. As depicted in Fig. 3d, nanozymes were deposited onto the disk electrode of a rotating ring-disk setup.36
 |
| | Fig. 3 (a) Time-dependent absorption change at 340 nm. (b) Mass spectra of the NADH oxidation reaction. (c) EPR spectra of the spin adducts formed from CYPMPO in the reaction of NADH and Ce1Pt. (d) RRDE measurement of the selective oxygen reduction of Ce1Pt in the O2-saturated electrolyte. (e) In situ ATR-FTIR spectra and (f) In situ Raman spectra of the nanozyme-catalyzed NADH oxidation reaction. | |
During the negative scan, the nanozyme acts as a catalyst, promoting the transfer of electrons from the electrode to O2 and catalyzing the ORR. The results indicate that both the disk current and ring current increase concurrently, suggesting the reduction of O2. The H2O2 yield (% H2O2) for Ce1Pt was measured at 0.58% with an electron transfer number of 3.99, confirming their 4e− pathways in the O2-saturated PBS buffer (Fig. S8a). HRP colorimetric assay also shows no color development of TMB, which confirms that the material prefers the four-electron pathway (Fig. S8b). Then, in situ attenuated total reflection FTIR (ATR-FTIR) spectroscopy and Raman spectroscopy were employed to monitor the NADH oxidation process in real time. As shown in Fig. 3e, the intensity of the C–N signals (∼1058 cm−1) and C–H signals (∼1395 cm−1) decreases significantly, while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N signals (∼1602 cm−1) progressively increase over time, indicating the oxidation of NADH. Furthermore, the gradually increasing peak intensities at ∼3200 cm−1 indicate the progressive formation of H2O. The emergence and increased intensity of the Raman band at ∼590 cm−1 during NADH oxidation are attributed to defect-induced lattice vibrational distortion in Ce-based oxides, which is associated with OVs. This observation confirms that OVs are dynamically engaged in the catalytic process (Fig. 3f).37
N signals (∼1602 cm−1) progressively increase over time, indicating the oxidation of NADH. Furthermore, the gradually increasing peak intensities at ∼3200 cm−1 indicate the progressive formation of H2O. The emergence and increased intensity of the Raman band at ∼590 cm−1 during NADH oxidation are attributed to defect-induced lattice vibrational distortion in Ce-based oxides, which is associated with OVs. This observation confirms that OVs are dynamically engaged in the catalytic process (Fig. 3f).37
DFT calculations of the catalytic process
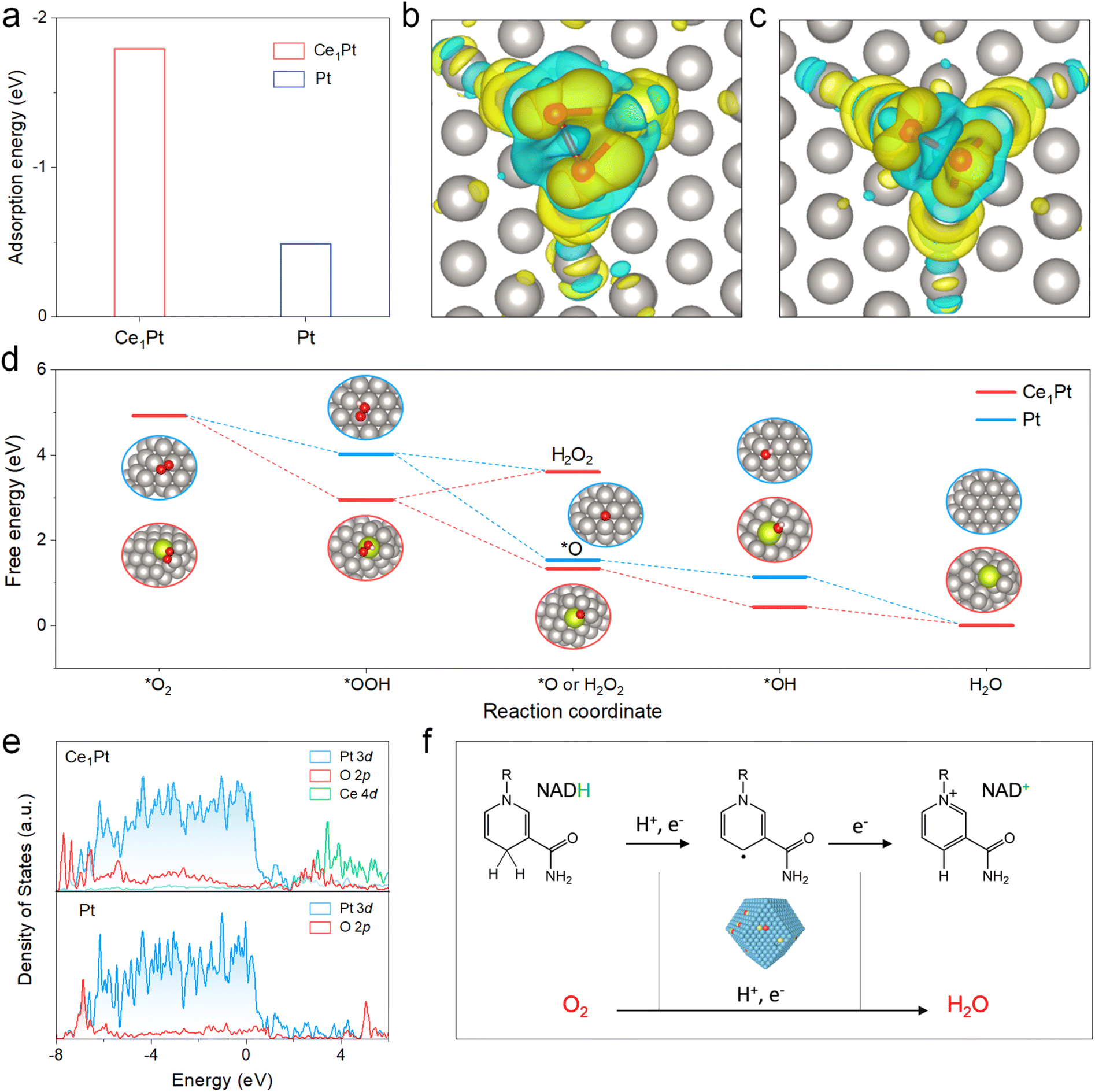
To gain deeper insight into the catalytic behavior of nanozymes, the mechanism was explored using density functional theory (DFT) calculations (Fig. S9). The adsorption energy of O2 on the OVs of Ce1Pt was calculated to be −1.80 eV, significantly lower than that of Pt (−0.49 eV), highlighting the superior adsorption capability of Ce1Pt (Fig. 4a). To investigate electron transfer between O2 and the nanozymes, charge density difference calculations were conducted (Fig. 4b and c, and S10). Upon O2 adsorption at the OVs, significant electron transfer was observed within the Pt and Ce sites, resulting in a decrease in electron density around the Pt or Ce atoms and an increase in electron density around the O2. As illustrated in Fig. 4d, the energy changes associated with the *OOH to *O step are calculated to be −2.68 eV for Ce1Pt and −2.49 eV for Pt. These values are notably lower than those of the *OOH to H2O2 step (0.66 eV for Ce1Pt and −0.42 eV for Pt), highlighting that the Ce1Pt nanozymes exhibit a strong preference for the 4e− pathway during O2 reduction. In addition, as shown in Fig. 4e, the d-band centers of both Ce1Pt and Pt are −2.31. While the d-band center of Ce1Pt remains unchanged, the introduction of Ce lowers the p-band center of the adsorbed O, resulting in greater overlap with the d-band center. This suggests that the incorporation of Ce enhances the interaction with oxygen intermediates, thereby promoting the oxygen reduction reaction. These findings indicate that Ce1Pt facilitates the dehydrogenation of NADH into a NAD˙ intermediate, followed by the transfer of electrons from NADH to O2, which is subsequently reduced to H2O (Fig. 4f).
 |
| | Fig. 4 (a) The absorption energy of nanozymes to O2. Calculated charge density differences to study the bonding interactions and the charge transfer of (b) Ce1Pt and (c) Pt among Pt, Ce, and O atoms. The yellow/blue isosurfaces denote an increase/decrease in electron density, and the grey, red, and yellow spheres represent Pt, O, and Ce atoms, respectively. (d) The free energy diagram of nanozymes determined by the DFT studies. (e) Projected DOS of Pt-d, Ce-d, and O-p orbitals of nanozymes. (f) Proposed NOX-like mechanism of Ce1Pt involving NADH oxidation and O2 reduction. | |
Practical sensing applications
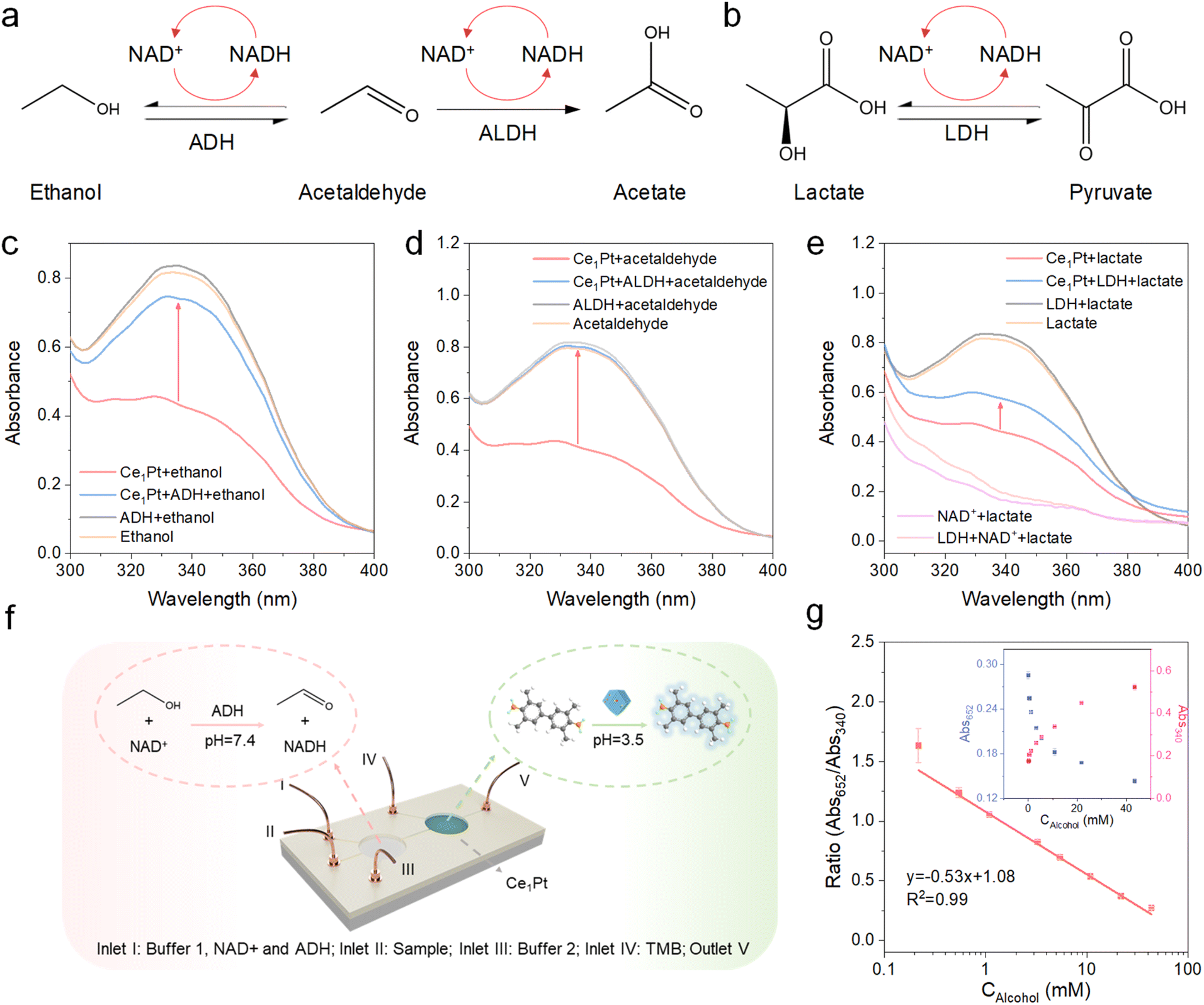
NAD+-dependent dehydrogenases in the human body, including alcohol dehydrogenase (ADH), aldehyde dehydrogenase (ALDH), and lactate dehydrogenase (LDH), drive substrate oxidation for metabolic homeostasis. However, NADH accumulation under substrate overload induces reverse catalysis, creating a vicious cycle of redox imbalance and pathological substrate retention (Fig. 5a and b).38–41 Especially for LDH, since most commercially available LDHs have higher reverse reaction activity, it becomes challenging to facilitate the forward reaction effectively.20 Therefore, we aim to leverage nanozymes for in situ coenzyme regeneration, which helps suppress the reverse reaction and restore metabolic balance. As shown in Fig. 5c, the characteristic peak of NADH at 340 nm is restored after the addition of ethanol and ADH. This demonstrates that the nanozyme can oxidize NADH to NAD+, as a coenzyme for ADH in the ethanol metabolism process. Similarly, the absorbance of NADH at 340 nm is also recovered upon the addition of acetaldehyde and ADH, or lactate and LDH (Fig. 5d and e). It is noteworthy that NAD+ alone cannot facilitate lactate dehydrogenation catalyzed by LDH, and the reaction occurs exclusively in the presence of both NADH and Ce1Pt. This is because the reverse reaction rate catalyzed by LDH in the human body is significantly higher than its forward reaction rate, which can only be driven under conditions where Ce1Pt continuously catalyzes the regeneration of NAD+. Proton nuclear magnetic resonance spectroscopy (1H NMR) confirmed that after the addition of Ce1Pt, ADH can effectively metabolize ethanol. When ethanol is used as the substrate, with ADH, ALDH, and the nanozyme added simultaneously, the metabolic product of the reaction is acetic acid (Fig. S11).42,43 Similarly, Ce1Pt also facilitates ALDH in metabolizing acetaldehyde and promotes LDH in converting lactate to pyruvate (Fig. S12).44,45 This demonstrates that the proposed NOX mimics exhibit the function of in situ coenzyme regeneration for inhibiting reverse reactions and promoting substrate metabolism. Furthermore, real-time assessment of in vivo metabolite concentrations offers a practical method for monitoring systemic health.46,47 This clinical relevance has accelerated the development of microfluidic platforms featuring integrated flow reactors, miniaturized architecture, and automated operation capability (Fig. 5f). The samples (ethanol), NAD+, ADH, and PBS buffer (pH 7.4) were introduced into the first reaction chamber for NADH production. Then, NaAc-HAc buffer (pH 3.0) was introduced to push the solution into the second reaction chamber and adjust the pH to around 3.5. The intrinsic oxidase-like activity of Ce1Pt induces competitive adsorption between TMB and NADH as substrates. As the blood alcohol concentration increases, the absorbance of the TMB-derived chromogenic product decreases. Based on this mechanism, we engineered a highly sensitive ratiometric sensor for quantifying NADH in blood ranging from 0.02–43 mM, with a low limit of detection of 0.012 mM mg 100 mL−1 (Fig. 5g).
 |
| | Fig. 5 (a and b) Schematic illustration of the ethanol, aldehyde, and lactate metabolic processes. Absorption spectra of NADH oxidation and regeneration reaction catalyzed by nanozymes with (c) ADH, (d) ALDH, or (e) LDH. (f) Nanozyme reaction system in a microfluidic device. (g) The ratiometric calibration curves for the detection of alcohol in blood. Inset: colorimetric response to different concentrations of alcohol in blood. | |
Conclusions
In summary, inspired by natural enzymes, we developed Ce1Pt nanozymes with NOX-like activity through the doping of atomic Ce. The introduction of Ce effectively reconstructed the Pt lattice and promoted the formation of OVs within the materials, and the NOX-like activity of the resultant Ce1Pt increased by 1.5 times that of Pt. Mechanistic studies reveal that Ce–OV sites enhance O2 adsorption, effectively reducing the reaction energy change and accelerating the reaction process. Moreover, the incorporation of Ce promotes the preference for the 4e− O2 reduction pathway in Ce1Pt, thereby reducing H2O2 generation and facilitating NADH oxidation. By harnessing its NOX-like activity, Ce1Pt can effectively facilitate in situ coenzyme regeneration, promoting the forward reaction of the enzyme-catalyzed reaction and thereby accelerating substrate metabolism. Finally, Ce1Pt was successfully utilized in fabricating a microfluidic chip for detecting alcohol in blood.
Ethical statement
All experiments were performed in accordance with the guidelines of the “Declaration of Helsinki” and approved by the ethics committee at “China Resources & Wisco General Hospital, Wuhan University of Science and Technology (Wuhan, P.R. China)”. Informed consents were obtained from human participants of this study. All human samples were de-identified of all identifying information. Informed and permitted consent was obtained from each subject in all clinical experiments of this manuscript.
Author contributions
Conceptualization, Y. T. and C. Z.; methodology, Y. T., Y. C., and P. Q.; investigation, Y. T., Y. C., R. L., W. J., and W. G.; writing – original draft, Y. T. and C. Z.; writing – review & editing, Y. T. and C. Z.; funding acquisition, W. G. and C. Z.; resources, H. S., W. G. and C. Z.; supervision, C. Z.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: SI methods, Fig. S1–S12 and Table S1. See DOI: https://doi.org/10.1039/d6sc00612d.
Acknowledgements
The authors gratefully acknowledge the financial support from the Fundamental Research Funds for the Central Universities (nos. CCNU25QN004 and CCNU24JCPT032), the Open Research Fund of the Key Laboratory of Ministry of Education, Hangzhou Normal University (no. KFJJ2023009), and the Program of Introducing Talents of Discipline to Universities of China (111 program, B17019). The authors acknowledge the support of the 1W1B beamline at the Beijing Synchrotron Radiation Facility for the XAFS experiments and China Resources & Wisco General Hospital, Wuhan University of Science and Technology, for providing serum samples in support of this study.
Notes and references
- L. Guarente, Science, 2016, 352, 1396–1397 Search PubMed.
- M. E. Migaud, M. Ziegler and J. A. Baur, Nat. Rev. Mol. Cell Biol., 2024, 25, 822–840 Search PubMed.
- D. V. Titov, V. Cracan, R. P. Goodman, J. Peng, Z. Grabarek and V. K. Mootha, Science, 2016, 352, 231–235 Search PubMed.
- R. P. Goodman, A. L. Markhard, H. Shah, R. Sharma, O. S. Skinner, C. B. Clish, A. Deik, A. Patgiri, Y.-H. H. Hsu, R. Masia, H. L. Noh, S. Suk, O. Goldberger, J. N. Hirschhorn, G. Yellen, J. K. Kim and V. K. Mootha, Nature, 2020, 583, 122–126 CrossRef CAS PubMed.
- X. Wang and H. H. P. Yiu, ACS Catal., 2016, 6, 1880–1886 Search PubMed.
- A. Patgiri, O. S. Skinner, Y. Miyazaki, G. Schleifer, E. Marutani, H. Shah, R. Sharma, R. P. Goodman, T.-L. To, X. Robert Bao, F. Ichinose, W. M. Zapol and V. K. Mootha, Nat. Biotechnol., 2020, 38, 309–313 CrossRef CAS PubMed.
- W. J. Quinn, J. Jiao, T. TeSlaa, J. Stadanlick, Z. Wang, L. Wang, T. Akimova, A. Angelin, P. M. Schäfer, M. D. Cully, C. Perry, P. K. Kopinski, L. Guo, I. A. Blair, L. R. Ghanem, M. S. Leibowitz, W. W. Hancock, E. K. Moon, M. H. Levine, E. B. Eruslanov, D. C. Wallace, J. A. Baur and U. H. Beier, Cell Rep., 2020, 33, 108500 Search PubMed.
- Y. Liu, J. Du, M. Yan, M. Y. Lau, J. Hu, H. Han, O. O. Yang, S. Liang, W. Wei, H. Wang, J. Li, X. Zhu, L. Shi, W. Chen, C. Ji and Y. Lu, Nat. Nanotechnol., 2013, 8, 187–192 Search PubMed.
- D. Zhu, M. Zhang, C. Wang, P. Gai and F. Li, Chem. Mater., 2022, 34, 11072–11080 Search PubMed.
- J. Chen, X. Zheng, J. Zhang, Q. Ma, Z. Zhao, L. Huang, W. Wu, Y. Wang, J. Wang and S. Dong, Natl. Sci. Rev., 2022, 9, nwab186 CrossRef CAS.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- C. Peng, R. Pang, J. Li and E. Wang, Adv. Mater., 2024, 36, 2211724 Search PubMed.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- J. Chen, X. Liu, G. Zheng, W. Feng, P. Wang, J. Gao, J. Liu, M. Wang and Q. Wang, Small, 2023, 19, 2205924 CrossRef CAS.
- J. Chen, Q. Ma, M. Li, D. Chao, L. Huang, W. Wu, Y. Fang and S. Dong, Nat. Commun., 2021, 12, 3375 Search PubMed.
- Q. Liu, A. Zhang, R. Wang, Q. Zhang and D. Cui, Nano-Micro Lett., 2021, 13, 154 Search PubMed.
- A. Rodriguez-Abetxuko, A. Reifs, D. Sánchez-deAlcázar and A. Beloqui, Angew. Chem., Int. Ed., 2022, 61, e202206926 Search PubMed.
- X. Liu, Z. Wan, K. Chen, Y. Yan, X. Li, Y. Wang, M. Wang, R. Zhao, J. Pei, L. Zhang, S. Sun, J. Li, X. Chen, Q. Xin, S. Zhang, S. Liu, H. Wang, C. Liu, X. Mu and X.-D. Zhang, Nano Lett., 2024, 24, 4924–4935 Search PubMed.
- J. Liu, B. Yu, M. Rong, W. Sun and L. Lu, Nano Today, 2024, 54, 102113 CrossRef CAS.
- D. Jiang, D. Ni, Z. T. Rosenkrans, P. Huang, X. Yan and W. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- L. Gao, H. Wei, S. Dong and X. Yan, Adv. Mater., 2024, 36, 2305249 Search PubMed.
- D. Li, J. He, G. Ding, Y. Xin, F. Feng, S. Ma, L. Lin, E. Wang and J. Wang, Adv. Healthcare Mater., 2025, 14, 2402785 Search PubMed.
- H. Rong, J. Mao, P. Xin, D. He, Y. Chen, D. Wang, Z. Niu, Y. Wu and Y. Li, Adv. Mater., 2016, 28, 2540–2546 Search PubMed.
- R. Feng, D. Li, H. Yang, C. Li, Y. Zhao, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2024, 36, 2309251 Search PubMed.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713–714 Search PubMed.
- G. Hou, Q. Wang, D. Xu, H. Fan, K. Liu, Y. Li, X.-K. Gu and M. Ding, Angew. Chem., Int. Ed., 2024, 63, e202402053 Search PubMed.
- X. Luo, H. Zhao, X. Tan, S. Lin, K. Yu, X. Mu, Z. Tao, P. Ji and S. Mu, Nat. Commun., 2024, 15, 8293 Search PubMed.
- F. Xu, Y. He, J. Zhang, G. Liang, C. Liu and J. Yu, Angew. Chem., Int. Ed., 2025, 64, e202414672 CrossRef CAS PubMed.
- W. Xu, H. Zhong, Y. Wu, Y. Qin, L. Jiao, M. Sha, R. Su, Y. Tang, L. Zheng, L. Hu, S. Zhang, S. P. Beckman, W. Gu, Y. Yang, S. Guo and C. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220315120 Search PubMed.
- Y. Wu, W. Xu, L. Jiao, Y. Tang, Y. Chen, W. Gu and C. Zhu, Mater. Today, 2022, 52, 327–347 Search PubMed.
- Y. Tang, Y. Chen, Y. Wu, W. Xu, Z. Luo, H.-R. Ye, W. Gu, W. Song, S. Guo and C. Zhu, Nano Lett., 2023, 23, 267–275 Search PubMed.
- S.-M. Jo, F. R. Wurm and K. Landfester, Angew. Chem., Int. Ed., 2021, 60, 7728–7734 Search PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS PubMed.
- Y. Tang, Y. Wu, W. Xu, L. Jiao, Y. Chen, M. Sha, H.-R. Ye, W. Gu and C. Zhu, Anal. Chem., 2022, 94, 1022–1028 Search PubMed.
- W. Jiang, Y. Wu, R. Su, W. Xu, W. Yang, Y. Qiu, Y. Cai, C. Wang, L. Hu, W. Gu and C. Zhu, Nano Lett., 2024, 24, 9635–9642 Search PubMed.
- Y. Tang, X. Liu, P. Qi, W. Xu, Y. Wu, Y. Cai, W. Gu, H. Sun, C. Wang and C. Zhu, Nano Lett., 2024, 24, 9974–9982 Search PubMed.
- Y. Huang, B. Long, M. Tang, Z. Rui, M.-S. Balogun, Y. Tong and H. Ji, Appl. Catal., B, 2016, 181, 779–787 Search PubMed.
- K. G. Hicks, A. A. Cluntun, H. L. Schubert, S. R. Hackett, J. A. Berg, P. G. Leonard, M. A. Ajalla Aleixo, Y. Zhou, A. J. Bott, S. R. Salvatore, F. Chang, A. Blevins, P. Barta, S. Tilley, A. Leifer, A. Guzman, A. Arok, S. Fogarty, J. M. Winter, H.-C. Ahn, K. N. Allen, S. Block, I. A. Cardoso, J. Ding, I. Dreveny, W. C. Gasper, Q. Ho, A. Matsuura, M. J. Palladino, S. Prajapati, P. Sun, K. Tittmann, D. R. Tolan, J. Unterlass, A. P. VanDemark, M. G. Vander Heiden, B. A. Webb, C.-H. Yun, P. Zhao, B. Wang, F. J. Schopfer, C. P. Hill, M. C. Nonato, F. L. Muller, J. E. Cox and J. Rutter, Science, 2023, 379, 996–1003 CrossRef CAS.
- H. Chen, Y. Li, H. Li, X. Chen, H. Fu, D. Mao, W. Chen, L. Lan, C. Wang, K. Hu, J. Li, C. Zhu, I. Evans, E. Cheung, D. Lu, Y. He, A. Behrens, D. Yin and C. Zhang, Nature, 2024, 631, 663–669 CrossRef CAS.
- J. Zhang, Y. Guo, X. Zhao, J. Pang, C. Pan, J. Wang, S. Wei, X. Yu, C. Zhang, Y. Chen, H. Yin and F. Xu, Nat. Rev. Cardiol., 2023, 20, 495–509 Search PubMed.
- A. Ghaddar, V. K. Mony, S. Mishra, S. Berhanu, J. C. Johnson, E. Enriquez-Hesles, E. Harrison, A. Patel, M. K. Horak, J. S. Smith and E. J. O'Rourke, Curr. Biol., 2023, 33, 1036–1046 Search PubMed.
- F. Gao, G. Liu, A. Chen, Y. Hu, H. Wang, J. Pan, J. Feng, H. Zhang, Y. Wang, Y. Min, C. Gao and Y. Xiong, Nat. Commun., 2023, 14, 6783 CrossRef CAS PubMed.
- H. Wang, H. Zheng, L. Ling, Q. Fang, L. Jiao, L. Zheng, Y. Qin, Z. Luo, W. Gu, W. Song and C. Zhu, ACS Nano, 2022, 16, 21266–21274 Search PubMed.
- E. T. Montrazi, K. Sasson, L. Agemy, A. Scherz and L. Frydman, Sci. Adv., 2024, 10, eadm8600 Search PubMed.
- Q. Wu, X. Liang, K. Wang, J. Lin, X. Wang, P. Wang, Y. Zhang, Q. Nie, H. Liu, Z. Zhang, J. Liu, Y. Pang and C. Jiang, Cell Metab., 2021, 33, 1988–2003e1987 Search PubMed.
- X. Wang, Y. Luo, K. Huang and N. Cheng, Adv. Agrochem., 2022, 1, 3–6 CrossRef.
- Y. Tang, Y. Wu, W. Xu, L. Jiao, W. Gu, C. Zhu, D. Du and Y. Lin, Adv. Agrochem., 2022, 1, 12–21 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Wenling
Gu
a and
Chengzhou
Zhu
c,
Wenling
Gu
a and
Chengzhou
Zhu