
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePushing the boundaries of BODIPY chemistry: 2-(dimethylamino)methyl BODIPYs as enablers of diversification with nucleophiles
Sergio
Serrano-Buitrago
 a,
Carla
Marcos
a,
Natalia
Casado
b,
Andrea
Aranda
a,
Florencio
Moreno
a,
Jorge
Bañuelos
b,
David
Valdivieso González
c,
Iván
López-Montero
cde,
Beatriz
L. Maroto
*a and
Santiago
de la Moya
*a
a,
Carla
Marcos
a,
Natalia
Casado
b,
Andrea
Aranda
a,
Florencio
Moreno
a,
Jorge
Bañuelos
b,
David
Valdivieso González
c,
Iván
López-Montero
cde,
Beatriz
L. Maroto
*a and
Santiago
de la Moya
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria S/N, Madrid 28040, Spain. E-mail: santmoya@ucm.es
bDepartamento de Química-Física, Universidad del País Vasco-EHU, Bilbao 48080, Spain
cDepartamento de Química Física, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria S/N, 28040 Madrid, Spain
dInstituto de Investigación Biomédica Hospital Doce de Octubre (imas12), Avda. de Córdoba S/N, 28041, Madrid, Spain
eInstituto Pluridisciplinar, Universidad Complutense de Madrid, P° Juan XXIII 1, 28040, Madrid, Spain
First published on 9th February 2026
Abstract
The BODIPY family of organic dyes has emerged as a versatile platform in photonic materials science, driven by their outstanding photophysical properties and the synthetic flexibility of BODIPY chemistry. Post-functionalization strategies have been pivotal in expanding BODIPY's application scope. However, current methodologies for rapid diversification with nucleophiles suffer from key limitations, including the use of unstable intermediates, limited substrate scope, and reliance on hazardous or costly reagents. In this work, we introduce 2-(dimethylamino)methyl BODIPYs as novel, stable, and easily accessible electrophilic intermediates that enable efficient BODIPY diversification with a broad range of neutral protic nucleophiles via unimolecular nucleophilic substitution. These intermediates are straightforwardly synthesized from highly common and accesible 2-unsubstituted BODIPYs through electrophilic aromatic substitution using inexpensive Eschenmoser's salt, and can be activated under mild conditions via simple amine quaternization. Importantly, this strategy is compatible with 3,5-dimethylated BODIPYs, preserving access to Knoevenagel-like BODIPY chemistry for large chromophore π-extension towarsds bathochromic shift into the red-to-NIR spectral region. The efficacy of this methodology is demonstrated through the synthesis of a number of BODIPY dyes with diverse substitution patterns and selectable photophysical behavior. Furthermore, we highlight its practical utility through the design of functional BODIPY derivatives, including ICT-based fluorescent pH indicators, fluorogenic acidotropic bioprobes, and water-soluble laser dyes.
Introduction
The BODIPY (boron-dipyrromethene) family of organic dyes has become a cornerstone in photonic research, driving innovation across a wide range of applications of high socio-economic relevance.1 This prominence stems from their exceptional photophysical behaviours, particularly their strong fluorescence, combined with their synthetic accessibility and versatile tunability via a set of well-established, straightforward organic-chemistry transformations collectively referred to as BODIPY chemistry.2Within the BODIPY-chemistry framework, post-functionalization strategies have proven particularly powerful for expanding the structural diversity and functional utility of BODIPY dyes.2f,k,m,o,r These strategies not only allow precise tuning of key photophysical properties, such as absorption and emission wavelengths or fluorescence efficiencies, but also enable the introduction of essential functional features, including water solubility,3 biorecognition capability for bioimaging and medical applications,4 or selective reactivity for chemical sensing.5 They also promote non-standard BODIPY photophysics, such as triplet-state population6 (relevant for photodynamic therapy,7 photon up-conversion,8 or photocatalysis9), efficient chiroptical responses of the inherently achiral BODIPY chromophore, including circularly polarized luminescence,10 or enhanced optoelectronic behaviours including OLED illumination,11 thereby significantly broadening the scope of BODIPY-based materials for a wide range of advanced photonic applications.
Since most common BODIPY post-functionalization strategies affect the chromophoric core, they can substantially alter the dye's photophysical properties.2f,k,m This presents a limitation when the goal is to introduce key functional moieties—such as for biorecognition—into a photophysically optimal BODIPY dye.4 In this context, the introduction of highly reactive functional groups, such as halogens, pseudohalogens, activated carboxyls, formyls, alkynes, or azides, at peripheral BODIPY positions (i.e., positions electronically isolated from the chromophore by an appropriate covalent spacer) is particularly attractive, as it generally preserves the dye's intrinsic photophysics.2f,k,m,4 However, spacer-based post-functionalization strategies, often involving BODIPY meso-phenyl or 3-styryl positions, typically require multi-step synthetic sequences, which can significantly reduce overall efficiency.2f,k,m,4
In this context, special attention should be given to the bimolecular nucleophilic substitution (SN2) reactions of 3-(bromomethyl) and 3,5-bis(bromomethyl) BODIPYs with neutral protic nucleophiles (NuH), particularly organic nucleophiles, as introduced by Ulrich and Ziessel in 2012 (Scheme 1).12 This methylene-spacer strategy offers rapid access to a wide range of asymmetric and symmetric BODIPY derivatives, since the key, reactive bromomethyl BODIPY intermediates are straightforwardly generated by electrophilic bromination of 3/5-methylated BODIPYs (enol-type BODIPY chemistry).12 However, the method seems to be restricted to sufficiently reactive heteroatom-based nucleophiles. Additionally, it seems to require electron-deficient (meso-iodoarylated) persubsituted BODIPYs,12 likely to activate the SN2 reaction and prevent BODIPY bromination by aromatic electrophilic substitution (SEAr). Moreover, the key bromomethylated intermediates (i.e., the electrophilic enablers of diversification with nucleophiles) are unstable and non-isolable, requiring bromination and substitution to be conducted sequentially in a one-pot protocol for each transformation (Scheme 1).12
 | ||
| Scheme 1 Comparison of previous methylene-spacer based methodologies for BODIPY post-functionalization with a neutral protic nucleophile (NuH), and the new approach reported herein. Key electrophilic enablers highlighted in boxes. | ||
To address these limitations, Martínez–Martínez, Chiara, Chiara and co-workers later proposed a more robust methylene-spacer strategy, which is based on 3-(acetoxymethyl) BODIPY as alternative, stable and isolable, electrophilic enabler.13 Notably, under Lewis acid activation, these compounds are proposed to undergo unimolecular nucleophilic substitution (SN1), enabling reaction with a significantly broader range of organic nucleophiles, including carbon-based ones (Scheme 1).13 However, this strategy requires starting from non-conventional 4,4-dicyano BODIPYs (CN-BODIPYs) instead of common 4,4-difluoro BODIPYs (F-BODIPYs), likely to prevent dye decomposition during the substitution step (and probably, competitive at-boron substitution of fluorines). Besides, it uses potentially hazardous reagents such as tin tetrachloride and trimethylsilyl cyanide to generate the key CN-BODIPY from the corresponding F-BODIPY, lead tetraacetate for achieving the required oxidative BODIPY-methyl acetoxylation, and the expensive, rare-earth-based scandium triflate catalyst to generate the key (BODIPY-3-yl)methyl cation (see Scheme 1).13 Moreover, it seems to require persubstituted BODIPYs, likely to prevent competitive SEAr reactions at the BODIPY core.13 Furthermore, it cannot be applied to certain nucleophiles, particularly amine-based nucleophiles likely due to their strong coordination with the required Lewis acid.13 It must be also noted here that the involved (BODIPY-3-yl)methyl cation must be significantly unstable owing to acidic nature of the BODIPY 3-methyl position. Together, these factors may substantially limit the practical applicability of this otherwise powerful functionalization method.
Additionally, using the BODIPY 3/5-methyls for enabling BODIPY post-functionalization with nucleophiles blocks valuable BODIPY-chromophore π-extension via Knoevenagel-type double condensation of 3,5-dimethyl BODIPYs with aromatic aldehydes, which is the most widely used strategy to easily achieve strong bathochromic shifts of the BODIPY absorption/emission bands.2d,4a,14 This is a big drawback in applications that require BODIPYs acting in the red-to-NIR region of the electromagnetic spectrum, such as certain bio-medical or optical communication applications.2d,4a,14
To address all these limitations, while keeping the advantage of the SN1 processes to enable BODIPY diversification with a broad range of neutral protic nucleophiles, we hypothesized on the alternative use of (BODIPY-2-yl)methyl cations. On the one hand, these carbocations should be more stable than the (BODIPY-3-yl)methyl counterparts and, therefore, they should be easier to generate. On the other hand, potential 3/5-methyl groups would remain available for enol-type BODIPY chemistry, particularly Knoevenagel-type double condensation for spectral red-shifting into the red-NIR region, to be performed either prior to or following the transformation.
To access these BODIPY carbocations, we propose exploiting the nucleophilic character of the BODIPY 2-position, which readily undergoes SEAr with a plethora or electrophiles, including C-electrophiles such as one-carbon synthetic equivalents (e.g., Vilsmeier–Haack reagent).15 In this context, we also hypothesized on the use of the inexpensive and commercially available Eschenmoser's salt (N-methyl-N-methylenemethanaminium iodide) as one-carbon electrophilic synthetic equivalent to directly generate 2-(dimethylamino)methyl BODIPYs from 2-unsusbtituted BODIPYs by SEAr. These intermediates could then be activated for the desired SN1 reaction with NuH via simple quaternization of the dimethylamino group.16 Nonetheless, the π-accelerated SN2 should not be discarded at the working reaction mechanism.
In this work, we demonstrate this possibility by reporting the effectiveness of 2-(dimethylamino)methyl BODIPYs as enablers of BODIPY diversification with an ample variety of NuHs under mild reaction conditions and without using hazardous and/or costly reagents or catalysts, showcasing some selected applications, particularly the development of valuable ICT-based BODIPY sensors (fluorescent pH indicators and fluorogenic acidotropic bioprobes) and water-soluble BODIPY laser dyes.
Results and discussion
Adjusting the methodology
To test our hypotheses, we selected: known 1,3,5,7-tetramethyl-8-(4-methylphenyl)-F-BODIPY (1) as the starting reactive BODIPY, standard quaternization with methyl iodide to activate the expected (dimethylamino)methyl-based enabler (2), and benzyl alcohol (BnOH) as the neutral nucleophile (Scheme 2).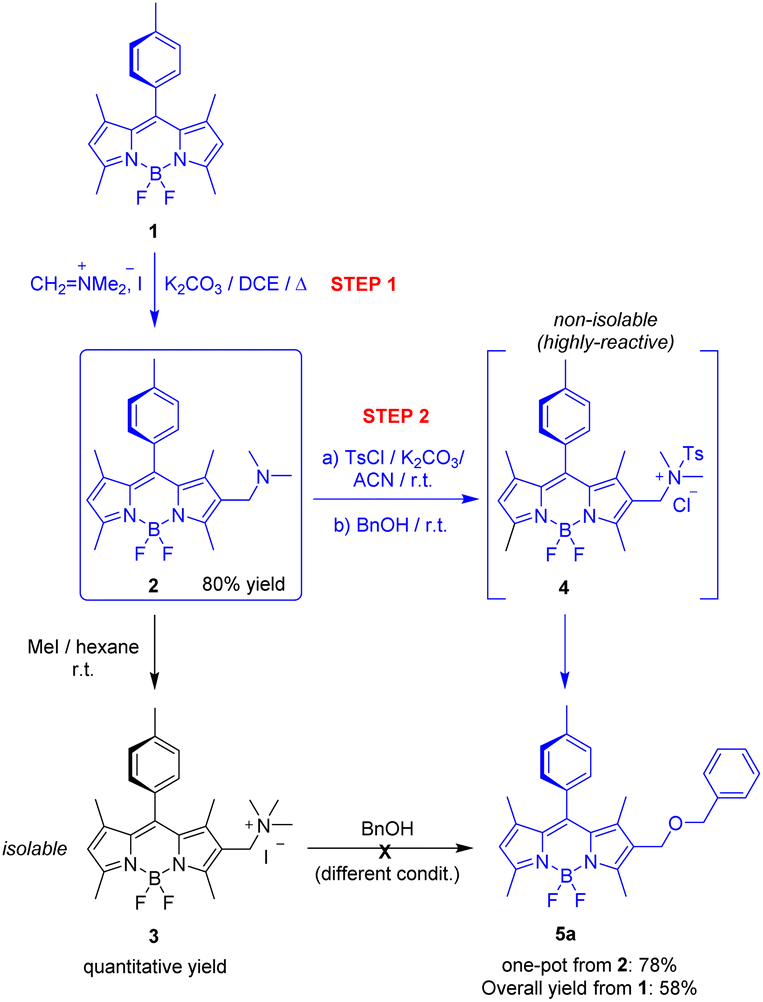 | ||
| Scheme 2 Chemoselective synthesis of 1-derived enabler 2; failed activation as the trimethylammonium salt 3; and successful activation as the (dimethyl)(tosyl)ammonium salt 4, which enables reaction with BnOH to generate 5. In blue, method's route. DCE: 1,2-dichloroethane; ACN: acetonitrile; Ts: tosyl; Bn: benzyl. See the SI for full experimental details. | ||
This selection was made based on the following criteria: (i) the accessibility of the starting materials; (ii) the absence of substitution at BODIPY positions 2 and 6 to probe chemoselectivity during enabler synthesis (mono-SEAr vs. bis-SEAr); (iii) the presence of substituents (methyls) at BODIPY positions 1 and 3 to evaluate potential steric hindrance effected near the reactive site of the enabler; (iv) the presence of methyls at BODIPY positions 3 and 5, enabling assessment of compatibility with enol-type BODIPY chemistry before or after functionalization.
As shown in Scheme 2, selected BODIPY 1 (synthesized according to the reported procedure17) underwent chemoselective aromatic substitution (mono-SEAr) with Eschenmoser's salt in the presence of potassium carbonate as base in refluxing 1,2-dichloroethane (DCE), affording derivative 2 in 80% yield. Subsequent standard methylation of 2 with methyl iodide in hexane led to the formation of the corresponding trimethylammonium salt in quantitative yield. However, this salt failed to undergo the expected nucleophilic substitution with BnOH under a variety of experimental conditions. Instead, recovery of the starting material, or dye decomposition under harder conditions, was consistently observed.
To address this issue, we explored the activation of 2 as the (dimethyl)(tosyl)ammonium salt 4. The new ammonium group was expected to serve as a better leaving group than the previous one owing to the strong electron-withdrawing nature of the tosyl ((4-methylphenyl)sulfonyl) moiety. Indeed, this activation—achieved via rapid reaction (<5 min) with tosyl chloride (TsCl) at room temperature—proved highly effective. It generated a highly reactive intermediate that could not be isolated from the reaction mixture, but reacted cleanly and rapidly (<3 min, r.t.) with BnOH to furnish BODIPY derivative 5a. This reactive behaviour strongly supports the in situ formation of the (dimethyl)(tosyl)ammonium salt (4), and the subsequent generation of the (BODIPY-2-yl)methyl cation by rapid release of N,N-dimethyltosylamide, which was isolated from the reaction mixture. It must be noted here that the π-accelerated SN2 mechanism cannot be ruled out based on these results. However, the notably rapid reaction kinetics observed for all the tested neutral nucleophiles, together with previous mechanistic reports on related BODIPY-based substitution reactions,13 support the proposed SN1 pathway.
As expected, (dimethylamino)methyl enabler 2 exhibited significantly lower fluorescence than the parent BODIPY 1 (Table 1), with the fluorescence quantum yield (ϕ) decreasing as the polarity of the medium increased (Tables S1 and S2 in the SI).
This behaviour is consistent with photoinduced intramolecular charge transfer (ICT) facilitated by the electron-donating amino group. Support for this mechanism comes from analogous trends observed in related aminomethyl BODIPY systems,12,13,18 the recovery of the starting fluorescent behaviour in the trimethylammonium derivative 3, and the intermediate photophysical behaviour of the (benzyloxy)methyl derivative 5a—consistent with the lower electron-donating ability of the oxygen in 5a compared to the amine nitrogen in 2—(see Table 1).
Thanks to its synthetic simplicity and efficiency, relatively low cost, and the absence of hazardous reagents, the two-step derivatization of 2-unsubstituted BODIPYs into methylene-spaced 2-substituted analogues—through the straightforward synthesis of a 2-[(dimethylamino)methyl]BODIPY enabler (see Scheme 2)—offers a practical and efficient method for diversifying conventional F-BODIPYs with organic nucleophiles.
Exploring the scope
 | ||
| Fig. 1 Workable diversification of compound 1via enabler 2 (see Scheme 2) with selected NuH species: benzyl alcohol (→ 5a), 2-bromoethanol (5b), 4-hydroxybenzaldehyde (5c), benzoic acid (5d), methoxyacetic acid (5e), piperidine (5f), propargylamine (5g), 3,4-dimethoxyaniline (5h), thiophenol (5i), and 2,4-dimethyl-3-ethylpyrrole (5j). The introduced methylene-spaced Nu moiety is highlighted in red (O-nucleophiles), blue (N-nucleophiles), green (S-nucleophile), and pink (C-nucleophile). Isolated chemical yields from 2 are indicated in parentheses. See the SI for full experimental details. | ||
It is important to note that our aim was not to test an exhaustive range of NuH species—since the intermediate (BODIPY-2-yl)methyl cation is expected to react with a wide variety of such nucleophiles, as has been previously reported for the analogous (BODIPY-3-yl)methyl cation.13 Instead, our goal was to evaluate: (i) the potential influence of steric hindrance imposed by the substituents located at BODIPY positions 1 and 3 (methyl groups in this case); (ii) possible competition from nucleophilic substitution at the BODIPY boron; (iii) compatibility with highly reactive functionalities such as primary alkyl halides or formyl groups; and (iv) the impact of the functionalization on the dye's photophysics.
To our pleasure, the isolated yields reached, which range from 84% for compound 5g (derivatization with highly nucleophilic propargylamine) to 44% for 5b (derivatization with highly unstable 2-bromoethanol), demonstrate the workability of the methodology with different NuHs, including highly reactive/unstable ones. These yields are comparable to those previously reported for analogous SN1 transformations involving (4,4-dicyanoBODIPY-3-yl)methyl cations,13 and are significantly influenced by the ability of the introduced moiety to act as a leaving group (nucleofuge), or undergo further transformation due to the presence of additional reactive groups.
 | ||
| Fig. 2 Workable diversification of selected BODIPYs 1 and 6–8 with benzyl alcohol (BnOH) via (dimethylamino)methylation (step 1) and subsequent, activated nucleophilic substitution (step 2) (see Scheme 2). See the SI for full experimental details. | ||
As shown in Fig. 2, the corresponding (dimethylamino)methylated enablers (2 and 9–11) were successfully obtained in all cases with comparable isolated yields, ranging from 70% for 11 to 85% for 9. This is particularly noteworthy considering the significantly varying reactivity of the selected BODIPYs toward SEAr (1 < 6 < 7 < 8), driven by the electronic effects of their substituents. These results can be attributed to the high electrophilic character of Eschenmoser's salt. Remarkably, even in the case of 7, where competitive Mannich-type condensation at the highly-reactive, enolizable-like meso-methyl position could occur,15d preferential mono-SEAr at the BODIPY 2-position was observed, confirming the retention of regioselectivity and chemoselectivity across the series.
Moreover, all the synthesized enablers underwent nucleophilic substitution with BnOH at the expected methylene position with similarly high yields (78–84%), with the exception of π-extended 11 that gave 14 with lower yield (51%) (Fig. 2). Nonetheless, the straightforward synthesis of compound 14 from π-extended BODIPY 8via enabler 11 demonstrates the applicability of our method for the late-stage functionalization of valuable 3,5-bis(styrylated) red-emitting BODIPYs with nucleophiles (Fig. 2).
Regarding valuable methylene-spaced amination at the BODIPY 2-position (e.g., compounds 2, 5f–h, and 9–11 in Fig. 1 and 2), the newly developed method provides an efficient alternative to the conventional Vilsmeier–Haack formylation followed by reductive amination.19 In contrast to this traditional route, which requires properly substituted F-BODIPY precursors, particularly 3-aryl-F-BODIPYs, to minimize side-product formation during formylation,15 the new strategy straightforwardly operates on a broad scope of starting materials.
 | ||
| Scheme 3 Workable double functionalization of BODIPY 1 with benzyl alcohol (BnOH) via (dimethylamino)methylation of 1-derived 2 (step 1*) and subsequent activated double nucleophilic substitution (step 2*). Experimental conditions were identical to those previously used for steps 1 and 2 (see Scheme 1), with only stoichiometric adjustment of the reagents in the case of step 2*. See the SI for full experimental details. | ||
Photophysical impact
To assess the photophysical impact of the developed BODIPY-functionalization method, we characterized all functionalized dyes—including the (dimethylamino)methylated enablers—and the corresponding starting materials in diluted solutions of organic solvents with varying polarity, specifically toluene, chloroform, and acetonitrile (ACN) (see the SI for full experimental details). This allowed us to investigate for potential solvent-dependent processes such as fluorescence-damping intramolecular charge transfer (ICT) or fluorescence-quenching photoinduced electron transfer (PET). The main photophysical data are detailed in the SI (see Tables S1, S3 and S4 in the SI).As shown in those tables, the developed methylene-spaced functionalization of BODIPYs exerts minimal influence on their light-absorption properties. This outcome was expected, as the methylene spacer likely prevents strong electronic coupling between the chromophoric BODIPY core and the appended n- or π-type substituents. Indeed, all the analysed BODIPY derivatives display sharp and intense absorption bands, with molar extinction coefficients (ε) ranging from 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 90000 M−1 cm−1 (e.g., see absorption spectra of representative green-emitting 2 and red-emitting 11 in Fig. 3). Moreover, the absorption maxima match with that of the corresponding starting BODIPY dye (∼500–515 nm for the derivatives of 1, 6, and 7, and ∼630 nm for those derived from π-extended 8; e.g., see the absorption spectra of 2 and 11 in Fig. 3).
000 to 90000 M−1 cm−1 (e.g., see absorption spectra of representative green-emitting 2 and red-emitting 11 in Fig. 3). Moreover, the absorption maxima match with that of the corresponding starting BODIPY dye (∼500–515 nm for the derivatives of 1, 6, and 7, and ∼630 nm for those derived from π-extended 8; e.g., see the absorption spectra of 2 and 11 in Fig. 3).
 | ||
| Fig. 3 Computed (CAM-B3LYP/6-311g*) electronic contour maps and energy levels (in eV) of the key frontier molecular orbitals (FMOs) for amine-based BODIPY 2, and related compounds 11 (bearing either styryls instead of methyls at 3/5) and 5h (bearing electron-richer amine (aniline) moiety), supporting feasibility for fluorescence-quenching reductive PET in the case of 5h. The absorption and fluorescence (shaded) spectra of 2 and 11 (top right, in toluene) and the fluorescence spectra of 2 and 5h (bottom right, in acetonitrile) are included for comparison purposes. For computed FMOs for related, PET-enabling 5j see Fig. S1 in the SI. See the SI for full experimental details. | ||
Functionalization with O- and S-nucleophiles generally maintains—or even slightly enhances—fluorescence quantum yields, regardless of solvent polarity (e.g., compare 1 with 5a–e, 5i–j and 16; 6 with 12; 7 with 13; and 8 with 14; in Fig. 4 and Table S1 in the SI).
 | ||
| Fig. 4 Representative structure- and solvent-dependent variations in fluorescence efficiency for selected BODIPYs (1 and 6–8; non-shaded data) upon methylene-spacer based functionalization with N-nucleophile (2, 5h and 9–11; blue-shaded data) and O-nucleophile (5a and 12–14; red-shaded data) moieties at BODIPY 2-position. Data selected from Tables S1, S3 and S4 in the SI. | ||
In contrast, substitution with N-based moieties leads to pronounced variations in fluorescence efficiency that are both structure- and solvent-dependent (e.g., compare 1 with 2 and 5h; 6 with 9; 7 with 10; and 8 with 11 in Fig. 4). Thus, the fluorescence of the derivatives bearing electron-rich tertiary-amine moieties (2, 5f, 15 and 9–11) clearly drops in polar solvents, where it becomes almost negligible (see Tables S1, S3 and S4 in the SI). This trend is softened when electron-poorer secondary-amine moieties are involved instead (e.g., compare 5f with 5g in Table S1 in the SI), or when the electron richness of the BODIPY core is enhanced (e.g., compare 2 with 11 in Fig. 4). As above mentioned, all this is consistent with polar-solvent promoted photoinduced ICT, taking place through the space from the electron-rich amino group to the electron-poor BODIPY core.21
Regarding fluorescence, the emission maxima wavelengths of the functionalized dyes are largely preserved relative to their respective starting materials (see Tables S1, S3 and S4 in SI). However, fluorescence intensity is highly dependent on the electronic nature of the introduced functionality (Fig. 4). The measured electrochemical properties of representative compounds (see the SI for full experimental details) further support the ICT pathway in those dyes functionalized with sufficiently electron-donating amine-based moieties (Fig. S2 in the SI). Thus, ICT-enabling 2 and 15, which bear tertiary alkyl amine groups, exhibit lower oxidation potentials (1.13 and 1.21 V, respectively) compared to related 5a (oxidation potential = 1.38 V), the latter bearing alcohol-based moiety and exhibiting no evidence of fluorescence-damping ICT.
Conversely, involving an electron-rich arenamine-based moiety strongly affects the fluorescence capability of the dye, as observed in the case of 5h (see Fig. 4), where almost null fluorescence is observed, even in the case of apolar toluene as the solvent. In this particular case, the strong electron-donating capability of the arenamine system, increased by the attached methoxyl groups, promotes fluorescence-quenching by PET from the electron-rich π-conjugated arenamine moiety to the electron-poor BODIPY core, as supported computationally (Fig. 3). Thus, light excitation is computed to promote electronic transition from HOMO−1 to LUMO, both localized at the BODIPY core, enabling thermodynamically-feasible reductive PET from the HOMO (located at the arenamine moiety) to the semivacant HOMO−1, which becomes fully occupied and prevents radiative deactivation by electronic transition from the semivacant LUMO (see Fig. 3). Indeed, compound 5h exhibits the lowest oxidation potential (0.78 V) among all the functionalized dyes analysed in this study, supporting its high propensity to undergo electron transfer processes (see Fig. S2 in the SI). A related fluorescence-quenching PET process, although less thermodynamically favoured, is also observed for compound 5j, which features an electron-rich 2-pyrrolyl moiety (see Fig. S1 in the SI).
All these results demonstrate that the developed functionalization methodology allows diversification of BODIPY dyes with a broad variety of NuHs without significantly affecting the outstanding light absorption capability of the starting dyes, which makes it ideal for developing dyed materials for photonic applications based on light absorption. Fluorescence maxima are also maintained, however, the starting BODIPY core and introduced nucleophilic moiety must be carefully selected when highly-brilliant final materials are targeted, and the use of electron-rich tertiary amines should be avoided in this case.
Showcasing some applications
The synthetic accessibility and selectable photophysical behaviour—achieved through the induction of ICT or PET processes—make the new BODIPY functionalization methodology ideally suited for the development of photonic applications in which designing ICT/PET-based fluorescence variations or the straightforward incorporation of additional functions is essential. To demonstrate this potential, we selected two representative application areas: ICT-based BODIPY probes,22 specifically fluorescent pH indicators and fluorogenic acidotropic bioprobes, and water-soluble BODIPY-based laser dyes.23,3cTo explore this potential, we selected compounds 2 and 5f (see Scheme 2 and Fig. 2), both featuring tertiary alkyl amine moieties, and monitored the changes in the fluorescence spectra in ethanol solution (4.0 µM) upon incremental addition of p-toluenesulfonic acid (TsOH). Gratifyingly, fluorescence intensity increased progressively with acid concentration, especially after the addition of 4 equivalents of TsOH (fluorescence quantum yield ≈ 0.05), reaching a plateau at approximately 7 equivalents (fluorescent quantum yield ≈ 0.65 for 5f, and ≈ 0.46 for 2) (see Fig. 5 for 5f, and S3 in the SI for 2). These results confirm that protonation of the amine effectively suppresses ICT, thereby significantly enhancing fluorescence efficiency. Notably, the fluorescent brightness of the protonated species in ethanol exceeds that of the non-protonated species in apolar solvents, and surpasses values reported for structurally related BODIPY-based fluorescent pH indicators featuring (dialkylamino)methyl substitution at the BODIPY 3-position instead of the BODIPY 2-position.13
 | ||
| Fig. 5 Variation of the fluorescence of compound 5f (4 µM in ethanol) upon incremental addition of TsOH (molar ratios from 1:0 to 1:20). Naked-eye brightness (top) and fluorescence spectra (bottom left). The dependence of the fluorescence intensity at 508 nm on proton concentration (bottom right) is also shown, highlighting pH sensitivity around apparent pH ≈ 4.5. See the SI for full experimental details. | ||
This pH-dependent fluorescence response is also evident in the fluorescence lifetimes, making possible advantageous lifetime-based pH sensing approaches.24 For instance, compound 5f shows a biexponential fluorescence decay in ethanol in its non-protonated form, with a dominant short lifetime component (∼0.15 ns). Upon protonation, the decay becomes monoexponential with a markedly longer lifetime of 3.20 ns—a 21-fold increase (see Fig. S4 in the SI).
The significant enhancement of fluorescence observed in 2-(dialkylamino)methylated BODIPYs upon protonation, combined with their straightforward synthesis, actuation pH range (∼4.0–5.0), expected biocompatibility and cell-membrane permeability, makes them ideal candidates as highly sensitive fluorogenic acidotropic lysosomal probes (Fig. 5).
Fluorogenic probes are especially valuable for bioimaging because they allow high-contrast visualization by minimizing background fluorescence in non-target regions.25 This “signal-on” behaviour not only improves imaging sensitivity but also simplifies experimental protocols by reducing the need for wash steps. In the context of live-cell imaging, where non-invasive, selective, and responsive indicators are key, fluorogenic probes capable of activation under specific physiological stimuli—such as pH shifts—are of particular interest for tracking intracellular events in real time.25
Particularly, cell-permeant pH-activatable (fluorogenic) acidotropic lysosomal probes (specifically marketed LysoSensors™) have been highlighted as valuable tools for investigating aberrant pH variations in lysosomes and other organelles, as well as alterations in lysosomal function or trafficking that are associated with major pathologies such as cancer, cystic fibrosis, and related diseases.26,27 However, pH-LysoSensor™ probes are limited to a small number of dyes, and their fluorescence efficiency in the protonated (fluorescent) state—and likely their long-term photostability—is significantly lower than that of the widely used LysoTracker™ probes, which are based on highly bright and photostable BODIPY fluorophores.26 Nevertheless, LysoTrackers™ do not exhibit marked fluorescence enhancement under acidic conditions,26 limiting their usefulness as ratiometric or pH-sensitive indicators. Consequently, the low fluorescence output of existing fluorogenic acidotropic sensors can pose a drawback in fluorescence-based applications, such as quantifying lysosomal number or tracking pH variations via flow cytometry or fluorometric analysis.26,28 In this context, the herein developed readily-accessible 2-(dialkylamino)methyl BODIPYs could surpass the performance of the commercially available LysoSensor™ probes.
To evaluate this potential, we selected compounds 2 and 5f and employed wild-type mouse embryonic fibroblasts (MEFs) as a cellular model. Computational prediction of pharmacokinetic parameters using the freely available SwissADME web tool29 indicated good permeability and biocompatibility profiles for both compounds. Encouragingly, both 2 and 5f efficiently stained living MEFs (see the SI for full experimental details), enabling fluorescence bioimaging of lysosomes with high specificity, as confirmed by co-localization with LysoTracker™ Red (pearson's coefficients > 0.6) (see Fig. S5 in the SI). This result supports pH-activatable fluorescence probing of lysosomes, as the selected dyes are poorly fluorescent under non-acidic conditions (e.g., see Fig. 5 for 5f).
To assess the superior behaviour of our fluorogenic acidotropic probes, we compared the fluorescence brightness of MEFs treated in parallel and under nearly identical conditions with commercial LysoTracker™ Green,26 and with compounds 2 and 5f. LysoTracker™ Green was selected due to its close structural and functional, biophotonic similarity to 2 and 5f: all three are highly bright, green-fluorescent BODIPY-based probes that exhibit comparable absorption and emission properties, and that accumulate specifically in lysosomes via a pendant tertiary alkyl amine that becomes protonated in the acidic lysosomal environment. However, as mentioned above, the fluorescence of LysoTracker™ Green is nearly pH-independent.
Satisfactorily, significantly less excitation laser power was required to achieve comparable imaging quality in the case of compound 2—and particularly compound 5f—when compared to LysoTracker™ Green (see Fig. 6, and S6 in the SI).
 | ||
| Fig. 6 Representative confocal laser scanning microscopy images of MEF cells acquired at a software setting of 20% laser power (corresponding to 274 µW power output), and represented in an 8-bit grayscale and pseudo-coloured using the LUT intensity saturation indicator. In this colour scale, fully saturated pixels (intensity = 255) are rendered red, and the darkest pixels (intensity = 0) are blue; intermediate intensities remain on a grayscale gradient. This colour scheme highlights overexposed regions in red and under-exposed/background areas in blue, allowing to compare signal distribution and saturation across samples for 2, 5f and LysoTracker™ Green when imaged at the same laser power. λexc = 488 nm and λem = 525 ± 25 nm. Scale bars are 20 µm. See the SI for full experimental details. | ||
Since fluorogenic acidotropic LysoSensors™ are significantly less fluorescent than LysoTrackers™, as mentioned above, all these results clearly demonstrate the higher performance of our 2-(dialkylamino)methyl BODIPYs as fluorogenic acidotropic lysosomal probes. Moreover, they highlight the potential of the newly developed BODIPY functionalization methodology to expand the current toolbox of acidotropic lysosomal sensors by enabling rapid access to advanced and structurally diverse probes, including valuable pH-activatable sensors operating in the biologically relevant red-to-NIR spectral region.30
In this context, we hypothesized that the straightforward generation of BODIPYs functionalized with highly hydrophilic groups, enabled by the methodology described herein, could offer a practical route to impart water solubility to otherwise hydrophobic 2/6-unsubstituted BODIPY dyes, thereby enabling their use in aqueous laser operation.
To test this hypothesis, we selected the trimethylammonium-functionalized BODIPY 3, along with the related difunctionalized dye 17 (Fig. 7), which was expected to exhibit improved aqueous solubility due to the presence of two ammonium groups. Compound 17 was prepared by quantitative methylation of the two amino groups of precursor 15 using methyl iodide under standard conditions.
 | ||
| Fig. 7 Molecular structure of the newly developed water-operable laser dye 17, shown alongside its normalized absorption (green-shaded), fluorescence (orange-shaded), and laser emission spectra (sharp red-shaded band with dotted line) recorded in pure water at high dye concentration (dye concentration = 0.2 mM). For comparison, the corresponding absorption and fluorescence spectra measured at dilute concentration (2 µM, dashed lines) in pure water are also included. See the SI for full experimental details. | ||
The water solubility of 3 and 17 was experimentally evaluated, along with their photophysical behaviour in pure water. Both dyes were found to be highly soluble and fully stable in water—particularly the doubly and symmetrically substituted dye 17, for which dye concentrations up to 0.2 mM could be achieved in pure water. Notably, no signs of aggregation were photophysically detected, even at the highest tested concentrations, as evidenced by the excellent overlap of the absorption and fluorescence spectra with those recorded at 100-fold lower concentrations (e.g., see Fig. 7 for 17). Importantly, the photophysical signatures recorded in pure water—such as a fluorescence quantum yield of approximately 0.50 for dye 17 (see Table S2 in the SI)—are comparable to those of the corresponding BODIPY precursor (e.g., precursor 1 in the case of 17) in organic solvents.
Remarkably, the high solubility, stability, and fluorescence of dye 17 in pure water enabled the recording of its laser emission in pure water. Thus, upon laser excitation of 17 in water, sharp and intense laser emission at 518 nm was observed (Fig. 7) with a lasing efficiency of 32%. This efficiency is comparable to that typically achieved by hydrophobic BODIPY laser dyes in organic solvents,33 clearly demonstrating the potential of the newly developed BODIPY functionalization methodology for advancing valuable organic laser dyes operating efficiently in pure water.
Conclusions
In summary, we have developed a novel and broadly applicable strategy for the diversification of common and readily accessible BODIPY dyes with nucleophiles, employing the corresponding 2-(dimethylamino)methyl-substituted dyes as stable, bench-accessible electrophilic intermediates. This platform enables efficient functionalization with a wide range of neutral protic nucleophiles under mild conditions. The methodology addresses longstanding limitations of related methylene-spacer based post-functionalization strategies, such as poor intermediate stability, narrow nucleophile and substrate scope, and reliance on hazardous or costly reagents.The developed new BODIPY post-functionalization method combines synthetic simplicity—relying on widely available 2-unsubstituted BODIPYs and relatively inexpensive commercial reagents—with operational convenience and potential scalability. Notably, its compatibility with 3,5-dimethyl BODIPY derivatives retains access to valuable red-to-NIR emissive dyes through well-established Knoevenagel-type double-condensation strategy for BODIPY spectral red-shifting.
Crucially, the method enables the controlled installation of diverse nucleophilic moieties—including those bearing reactive functionalities—across a broad structural space, while preserving the intrinsic absorption properties of the parent BODIPY core. Fluorescence efficiency is also maintained when the introduced groups are carefully selected, avoiding quenching elements such as electron-rich tertiary amines.
The demonstrated ability to generate a wide array of BODIPY dyes with tailored substitution patterns and selectable photophysical behaviour opens new avenues for the design of functional, fully organic molecular photonic tools. This potential is exemplified by the successful development of advanced fluorescent pH indicators, water-soluble laser dyes, and fluorogenic acidotropic bioprobes.
Altogether, the post-functionalization methodology presented here significantly expands the synthetic toolbox of BODIPY chemistry, and establishes a versatile foundation for future innovation in the fields of organic photonics and chiroptics.
Author contributions
S. d. l. M., conceptualization; S. d. l. M. and B. L. M. supervision; S. S.-B., C. M. and A. A., investigation (synthetic development); F. M., investigation (structural characterization); N. C. and J. B., investigation ((photo)physical behaviour); D. V. G. and I. L.-M., investigation (live-cell imaging validation); B. L. M., S. d. l. M., J. B., F. M. and I. L.-M., writing (review and editing); S. d. l. M. and B. L. M., writing (original draft).Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general methods and equipment, synthetic procedures and characterization, NMR spectra of new compounds, as well as additional figures concerning (photo)physical and (bio)photonical behaviours. See DOI: https://doi.org/10.1039/d5sc08677a.Acknowledgements
S. d. l. M. and B. L. M. acknowledge project PID2024-157648-NB-C21 (granted by MICIU/AEI/10.13039/501100011033 and FEDER, UE). J. B. acknowledges projects PID2024-157648-NB-C22 (granted by MICIU/AEI/10.13039/501100011033 and FEDER, UE) and IT2001-26 (granted by Basque Government). S. S.-B., C. M. and D. V. G. acknowledge UCM and Banco Santander for their pre-doctoral contracts (CT58/21-CT59/21, CT25/24 and CT58/21-CT59/21, respectively).Notes and references
- (a) X. Hu, Z. Fang, C. Zhu, Y. Yang, Z. Yang and W. Huang, Adv. Funct. Mater., 2024, 34, 2401325 CrossRef CAS; (b) X. Hu, Z. Fang, C. Zhu, Y. Yang, Z. Yang and W. Huang, Adv. Funct. Mater., 2024, 34, 2401325 CrossRef; (c) P. Stachelek, S. Serrano-Buitrago, B. L. Maroto, R. Pal and S. de la Moya, ACS Appl. Mater. Interfaces, 2024, 16, 67246–67254 CrossRef PubMed; (d) J. Chen, Q. Sha, M. U. Rehman, M. Wu, Z. Hu and F. Wu, ACS Appl. Nano Mater., 2024, 7, 23122–23132 CrossRef; (e) A. Garcia-Sampedro, A. Prieto-Castañeda, A. R. Agarrabeitia, J. Bañuelos, I. Garcia-Moreno, A. Villanueva, S. de la Moya, M. J. Ortiz and P. Acedo, J. Mater. Chem. B, 2024, 12, 7618–7625 RSC; (f) H. Li, X. Xu, R. Guan, A. Movsesyan, Z. Lu, Q. Xu, Z. Jiang, Y. Yang, M. Khan, J. Wen, H. Wu, S. de la Moya, G. Markovich, H. Hu, Z. Wang, Q. Guo, T. Yi, A. O. Govorov, Z. Tang and X. Lan, Nat. Commun., 2024, 15, 4846 CrossRef; (g) L. Gutierrez-Galvez, E. Enebral-Romero, M. A. Valle Amores, C. Pina Coronado, I. Torres, D. Lopez-Diego, M. Luna, A. Fraile, F. Zamora, J. Aleman, J. Álvarez, M. J. Capitán, E. Lorenzo and T. García-Mendiola, Nanoscale, 2025, 17, 8126–8140 RSC; (h) Z. Chen, Y. Zhou, L. Li, W. Ma, Y. Li and Z. Yang, Small, 2025, 21, 2411787 Search PubMed; (i) N. Lin and T. Mani, J. Am. Chem. Soc., 2025, 147, 7187–7190 Search PubMed; (j) F. Wang, F. Tan, W. Zhao, S. Zhou, Q. Xu, L. Kan, L. Zhu, P. Gu and J. Lu, Angew. Chem., Int. Ed., 2025, 64, e202425017 Search PubMed.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef PubMed; (b) R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC; (c) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef; (d) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC; (e) J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC; (f) N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org Chem., 2015, 2015, 6577–6595 CrossRef; (g) V. Lakshmi, M. R. Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501–2517 RSC; (h) L. Jean-Gerard, W. Vasseur, F. Scherninski and B. Andrioletti, Chem. Commun., 2018, 54, 12914–12929 RSC; (i) E. Bodio and C. Goze, Dyes Pigm., 2019, 160, 700–710 Search PubMed; (j) R. G. Clarke and M. J. Hall, Adv. Heterocycl. Chem., 2019, 128, 181–261 CrossRef; (k) N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 Search PubMed; (l) N. A. Bumagina, E. V. Antina, A. A. Ksenofontov, L. A. Antina, A. A. Kalyagin and M. B. Berezin, Coord. Chem. Rev., 2022, 469, 214684 Search PubMed; (m) R. L. Gapare and A. Thompson, Chem. Commun., 2022, 58, 7351–7359 RSC; (n) D. Alvarez-Gutierrez, D. Sampedro, M. C. Jimenez, R. Perez-Ruiz and A. C. S. Appl, Opt. Mater., 2024, 2, 1780–1789 Search PubMed; (o) M. Bogomolec, M. Glavas and I. Skoric, Molecules, 2024, 29, 5157 CrossRef PubMed; (p) J. G. Becerra-Gonzalez, E. Peña-Cabrera and J. L. Belmonte-Vazquez, Tetrahedron, 2024, 168, 134334 Search PubMed; (q) R. Segovia-Perez, M. Ibarra-Rodriguez, B. M. Munoz-Flores and V. M. Jimenez-Perez, Dyes Pigm., 2025, 239, 112740 Search PubMed; (r) A. S. Sherudillo, L. A. Antina and E. V. Antina, Coord. Chem. Rev., 2025, 545, 217030 CrossRef.
- (a) C. S. Mahanta, V. Ravichandiran and S. P. Swain, ACS Appl. Bio Mater., 2023, 6, 2995–3018 CrossRef PubMed; (b) H.-B. Cheng, X. Cao, S. Zhang, K. Zhang, Y. Cheng, J. Wang, J. Zhao, L. Zhou, X.-J. Liang and J. Yoon, Adv. Mater., 2023, 35, 2207546 CrossRef; (c) C. Schad, C. Ray, C. Díaz-Norambuena, S. Serrano-Buitrago, F. Moreno, B. L. Maroto, I. García-Moreno, M. Muñoz-Úbeda, I. López-Montero, J. Bañuelos and S. de la Moya, Chem. Sci., 2025, 16, 8030–8039 RSC.
- (a) Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC; (b) T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 Search PubMed; (c) S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121–134 Search PubMed; (d) V.-N. Nguyen, J. Ha, M. Cho, H. Li, K. M. K. Swamy and J. Yoon, Coord. Chem. Rev., 2021, 439, 213936 CrossRef; (e) S. Samanta, K. Lai, F. Wu, Y. Liu, S. Cai, X. Yang, J. Qu and Z. Yang, Chem. Soc. Rev., 2023, 52, 7197–7726 RSC; (f) H. Ahmad, S. Muhammad, M. Mazhar, A. Farhan, M. S. Iqbal, H. Hiria, C. Yu, Y. Zhang and B. Guo, Coord. Chem. Rev., 2025, 526, 216383 CrossRef; (g) P. P. P. Kumar, S. Saxena and R. Joshi, Colorants, 2025, 4, 13 CrossRef; (h) C. Kalarikkal and C. A. Swamy, J. Mater. Chem. B, 2025, 13, 12831–12868 RSC.
- (a) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 Search PubMed; (b) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC; (c) Y. T. Nguyen, S. Shin, K. Kwon, N. Kim and S. W. Bae, J. Chem. Res., 2023, 47, 17475198231168961 CrossRef; (d) N. A. Bumagina and E. V. Antina, Coord. Chem. Rev., 2024, 505, 215688 CrossRef; (e) S. Routray, S. Acharya, L. Nayak, S. Pattnaik and R. Satapathy, RSC Adv., 2025, 15, 9910–9951 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- (a) S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC; (b) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC; (c) C. S. Kue, S. Y. Ng, S. H. Voon, A. Kamkaew, L. Y. Chung, L. V. Kiew and H. B. Lee, Photochem. Photobiol. Sci., 2018, 17, 1691–1708 CrossRef; (d) A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef; (e) M. L. Agazzi, M. B. Ballatore, A. M. Durantini, E. N. Durantini and A. C. Tomé, J. Photochem. Photobiol., C, 2019, 40, 21–48 CrossRef; (f) R. Prieto-Montero, A. Prieto-Castañeda, R. Sola-Llano, A. R. Agarrabeitia, D. García-Fresnadillo, I. López-Arbeloa, Á. Villanueva, M. J. Ortiz, S. de la Moya and V. Martínez-Martínez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef PubMed; (g) X. Hu, Z. Fang, C. Zhu, Y. Yang, Z. Yang and W. Huang, Adv. Funct. Mater., 2024, 34, 2401325 CrossRef.
- D. Alvarez-Gutierrez, D. Sampedro, M. C. Jimenez and R. Perez-Ruiz, ACS Appl. Opt. Mater., 2024, 2, 1780–1789 Search PubMed.
- (a) P. De Bonfils, L. Peault, P. Nun and V. Coeffard, Eur. J. Org Chem., 2021, 2021, 1809–1824 CrossRef; (b) P. Rana, N. Singh, P. Majumdar and S. P. Singh, Coord. Chem. Rev., 2022, 470, 214698 CrossRef; (c) D. Zhang, L. Liu, X. Zhang, J. Lu and X.-D. Jiang, Resour. Chem. Mater., 2024, 3, 103–122 CAS.
- (a) H. Lu, J. Mack, T. Nyokong, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2016, 318, 1–15 Search PubMed; (b) H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS; (c) M. J. Hall and S. de la Moya, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer, Singapore, 2020, pp. 117–150 Search PubMed; (d) M. Ikeshita and T. Tsuno, Phys. Chem. Chem. Phys., 2025, 27, 17116–17129 Search PubMed.
- (a) A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3400 RSC; (b) S. P. Singh and T. Gayathri, Eur. J. Org Chem., 2014, 2014, 4689–4707 CrossRef CAS; (c) D. Ho, R. Ozdemir, H. Kim, T. Earmme, H. Usta and C. Kim, ChemPlusChem, 2019, 84, 18–37 CrossRef CAS; (d) M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS; (e) B. M. Squeo and M. Pasini, Supramol. Chem., 2020, 32, 56–70 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Haefele, J. Org. Chem., 2012, 77, 4298–4311 CrossRef CAS.
- A. Blázquez-Moraleja, L. Maierhofer, E. Mann, R. Prieto-Montero, A. Oliden-Sánchez, L. Celada, V. Martínez-Martínez, M.-D. Chiara and J. L. Chiara, Org. Chem. Front., 2022, 9, 5774–5789 RSC.
- (a) H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC; (b) J. Jiménez, C. Díaz-Norambuena, S. Serrano, S. C. Ma, F. Moreno, B. L. Maroto, J. Bañuelos, G. Muller and S. de la Moya, Chem. Commun., 2021, 57, 5750–5753 RSC; (c) C. Kalarikkal and P. C. A. Swamy, J. Mater. Chem. B, 2025, 13, 12831–12868 RSC; (d) Y. Li, M. Jiang, M. Yan, J. Ye, Y. Li, W. Dehaen and S. Yin, Coord. Chem. Rev., 2024, 506, 215718 CrossRef CAS.
- (a) L. Jiao, C. Yu, J. Li, Z. Wang, M. Wu and E. Hao, J. Org. Chem., 2009, 74, 7525–7528 CrossRef CAS PubMed; (b) C. Yu, L. Jiao, H. Yin, J. Zhou, W. Pang, Y. Wu, Z. Wang, G. Yang and E. Hao, Eur. J. Org Chem., 2011, 2011, 5460–5468 CrossRef CAS; (c) D. E. Ramirez-Ornelas, E. Alvarado-Martinez, J. Bañuelos, I. Lopez Arbeloa, T. Arbeloa, H. M. Mora-Montes, L. A. Perez-Garcia and E. Pena-Cabrera, J. Org. Chem., 2016, 81, 2888–2898 CrossRef CAS; (d) E. Palao-Utiel, L. Montalvillo-Jimenez, I. Esnal, R. Prieto-Montero, A. R. Agarrabeitia, I. Garcia-Moreno, J. Bañuelos, I. Lopez-Arbeloa, S. de la Moya and M. J. Ortiz, Dyes Pigm., 2017, 141, 286–298 CrossRef CAS.
- (a) Z.-X. Wang and B. Yang, Org. Biomol. Chem., 2020, 18, 1057–1072 RSC; (b) J. Templ and M. Schnürch, Chem. – Eur. J., 2024, 30, e202400675 Search PubMed.
- A. Cui, X. Peng, J. Fan, X. Chen, Y. Wu and B. Guo, J. Photochem. Photobiol., A, 2007, 186, 85–92 Search PubMed.
- E. Palao, S. de la Moya, A. R. Agarrabeitia, I. Esnal, J. Bañuelos, I. López-Arbeloa and M. J. Ortiz, Org. Lett., 2014, 16, 4364–4367 CrossRef CAS PubMed.
- (a) X. Gu, C. Liu, Y.-C. Zhu and Y.-Z. Zhu, Tetrahedron Lett., 2011, 52, 5000–5003 CrossRef CAS; (b) C. Zhao, Y. Zhang, P. Feng and J. Cao, Dalton Trans., 2012, 41, 831–838 Search PubMed; (c) L. Wang, Y. Xiao, W. Tian and L. Deng, J. Am. Chem. Soc., 2013, 135, 2903–2906 CrossRef CAS PubMed; (d) C. Zhao, X. Li and J. Zhang, Inorg. Chim. Acta, 2014, 412, 32–37 CrossRef CAS; (e) H. Wang, Y. Wu, Y. Shi, P. Tao, X. Fan, X. Su and G.-C. Kuang, Chem. – Eur. J., 2015, 21, 3219–3223 CrossRef CAS; (f) Z. Xu, Y. Huang, Y. Cao, T. Jin, K. A. Miller, A. L. Kaledin, D. G. Musaev, T. Lian and E. Egap, J. Chem. Phys., 2020, 153, 154201 CrossRef; (g) A. M. Gómez, A. H. G. David, A. G. Campaña, J. M. Cuerva, L. Díaz-Casado, C. Uriel, A. Oliden-Sánchez, J. Bañuelos, I. García-Moreno, L. Infantes, C. M. Cruz, J. T. Chambi and J. C. López, J. Org. Chem., 2024, 89, 18522–18528 CrossRef.
- (a) I. García-Moreno, L. Wang, Á. Costela, J. Bañuelos, I. López Arbeloa and Y. Xiao, ChemPhysChem, 2012, 13, 3923–3931 CrossRef; (b) E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. de la Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef; (c) E. Avellanal-Zaballa, J. Ventura, L. Gartzia-Rivero, J. Bañuelos, I. García-Moreno, C. Uriel, A. M. Gómez and J. C. López, Chem. – Eur. J., 2019, 25, 14959–14971 CrossRef CAS PubMed.
- (a) R. B. Alnoman, P. Stachelek, J. G. Knight, A. Harriman and P. G. Waddell, Org. Biomol. Chem., 2017, 15, 7643–76513 RSC; (b) F.-Z. Li, Z. Wu, C. Lin, Q. Wang and G.-C. Kuang, Results Chem., 2022, 4, 1000384 Search PubMed.
- Y. Wu, C. Ge, Y. Zhang, Y. Wang and D. Zhang, Front. Chem., 2023, 11, 1304531 CrossRef CAS PubMed.
- S. Sinha, A. K. Ray, S. Kundu, S. Sasikumar and K. Dasgupta, Appl. Phys. B, 2002, 75, 85 CrossRef CAS.
- (a) A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387–6395 CrossRef CAS PubMed; (b) Y. Ning, S. Cheng, J.-X. Wang, Y.-W. Liu, W. Feng, F. Li and J.-L. Zhang, Chem. Sci., 2019, 10, 4227–4235 Search PubMed; (c) J. J. Rennick, C. J. Nowell, C. W. Pouton and A. P. R. Johnston, Nat. Commun., 2022, 13, 6023 CrossRef CAS PubMed.
- (a) J. B. Grimm, L. M. Heckman and L. D. Lavis, Prog. Mol. Biol. Transl. Sci., 2013, 113, 1–34 CAS; (b) A. Nadler and C. Schultz, Angew. Chem., Int. Ed., 2013, 52, 2408–2410 CrossRef CAS; (c) A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366–375 Search PubMed; (d) D. Si, Q. Li, Y. Bao, J. Zhang and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e20230764 Search PubMed.
- Probes for Lysosomes, Peroxisomes and Yeast Vacuoles—Section 12.3, in The Molecular Probes Handbook, ed. M. T. Z. Spence and I. D. Johnson, Thermo Fisher Scientific, 11th edn, 2010. Search PubMed.
- (a) S. S. Chauhan, X.-J. Liang, A. W. Su, A. Pai-Panandiker, D.-W. Shen, J. A. Hanover and M. M. Gottesman, Br. J. Cancer, 2003, 88, 1327–1334 Search PubMed; (b) N. Raben, L. Shea, V. Hill and P. Plotz, Methods Enzymol., 2009, 453, 417–449 Search PubMed; (c) P. M. Haggie and A. S. Verkman, J. Biol. Chem., 2009, 284, 7681–7686 Search PubMed; (d) D. M. Wolfe, J.-H. Lee, A. Kumar, S. Lee, S. J. Orenstein and R. A. Nixon, Eur. J. Neurosci., 2013, 37, 1949–1961 CrossRef PubMed; (e) J. Zeng, O. S. Shirihai and M. W. Grinstaff, J. Life Sci., 2020, 2, 25–37 Search PubMed.
- (a) S. Chow and D. Hedley, Curr. Protoc. Cytom., 2001, 9 Search PubMed , PMID: 18770756; (b) L. Ma, Q. Ouyang, G. C. Werthmann, H. M. Thompson and E. M. Morrow, Front. Cell Dev. Biol., 2017, 5, 71 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 Search PubMed.
- (a) G. K. Vegesna, J. Janjanam, J. Bi, F.-T. Luo, J. Zhang, C. Olds, A. Tiwari and H. Liu, J. Mater. Chem. B, 2014, 2, 4500–4508 Search PubMed; (b) A. Mukherjee, P. C. Saha, R. S. Das, T. Bera and S. Guha, ACS Sens., 2021, 6, 2141–2146 Search PubMed; (c) A. Mukherjee, P. C. Saha, S. Kar, P. Guha, R. S. Das, T. Bera and S. Guha, ChemBioChem, 2023, 24, e20220064 Search PubMed; (d) Z. Li, S. Yang, H. Xiao, Q. Kang, N. Li, G.-L. Wu, S. Tan, W. Wang, Q. Fu, X. Tang, J. Zhou, Y. Huang, G. Chen, X. Tan and Q. Yang, Bioconjugate Chem., 2024, 35, 1015–1023 CrossRef PubMed.
- (a) O. G. Peterson, S. A. Tuccio and B. B. Snavely, Appl. Phys. Lett., 1970, 17, 245–247 CrossRef; (b) B. Wellegehausen, L. Laepple and H. Welling, Appl. Phys., 1975, 6, 335 CrossRef; (c) H. El-Kashef, Physica B, 2000, 279, 295 CrossRef; (d) S. Sinha, A. Ray and K. Dasgupta, J. Appl. Phys., 2000, 87, 3222 CrossRef; (e) H. El-Kashef, Physica B, 2002, 311, 376 CrossRef; (f) S. Sinha, A. K. Ray, S. Kundu, S. Sasikumar and K. Dasgupta, Appl. Phys. B, 2002, 75, 85 CrossRef; (g) A. Sen, A. K. Mora, S. K. Agarwalla, G. Sridhar, S. Kundu and S. Nath, Spectrochim. Acta, Part A, 2022, 282, 121642 CrossRef PubMed.
- Laser & Fluorescent Dyes, Luxottica/Exciton, https://exciton.luxottica.com/laser-dyes.html, accessed 2024-10-26.
- E. Avellanal-Zaballa, L. Gartzia-Rivero, T. Arbeloa and J. Bañuelos, Int. Rev. Phys. Chem., 2022, 41, 177–203 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |