
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStealing from a distant neighbor: an unexpectedly fast long-span peroxy radical hydrogen-shift reaction in a long-chain diether
Hongmin
Yu
 a,
Thomas
Golin Almeida
b,
Samir P.
Rezgui
c,
Vili-Taneli
Salo
b,
John D.
Crounse
a,
Brian M.
Stoltz
c,
Henrik G.
Kjaergaard
b and
Paul O.
Wennberg
*ad
a,
Thomas
Golin Almeida
b,
Samir P.
Rezgui
c,
Vili-Taneli
Salo
b,
John D.
Crounse
a,
Brian M.
Stoltz
c,
Henrik G.
Kjaergaard
b and
Paul O.
Wennberg
*ad
aDivision of Geological and Planetary Sciences, California Institute of Technology, Pasadena, CA, USA. E-mail: wennberg@caltech.edu
bDepartment of Chemistry, University of Copenhagen, Copenhagen, Denmark
cDivision of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, USA
dDivision of Engineering and Applied Science, California Institute of Technology, Pasadena, CA, USA
First published on 16th March 2026
Abstract
Gas-phase autoxidation is an atmospheric chemistry reaction mechanism capable of transforming volatile organic compounds (VOCs) into highly oxygenated organic molecules (HOMs) that contribute to secondary organic aerosol (SOA) formation and growth. The key steps in this mechanism are intramolecular hydrogen shift (H-shift) reactions in organic peroxy radicals (RO2). For acyclic saturated molecules, these H-shift reactions are generally sufficiently slow that they cannot compete with atmospheric bimolecular reactions with NOx species, except for the 1,5 and 1,6 H-shifts, occurring via transition states (TS) of six- and seven-membered rings. Here, we report a surprisingly fast long-range H-shift reaction in a RO2 formed in the photo-oxidation of a volatile diether. In 1,2-diethoxyethane (1,2-DEE), we observe experimentally a 1,8 H-shift reaction that occurs with a rate coefficient on the order of ∼1 s−1 at 294 K – a rate that outcompetes all other RO2 unimolecular chemistry in the system and will, under most atmospheric conditions, outcompete bimolecular processes as well. Theoretical calculations indicate that activation of the C–H bond by an α-oxyl substituent and weaker transannular strain in the 1,8 H-shift transition state, combined with inductive deactivation of C–H bonds by a β-oxyl group at the abstraction site of competing 1,5 and 1,6 H-shifts, enable the longer-span 1,8 H-shift to be competitive. Our findings broaden the recognized reactivity of functionalized RO2 and highlight the potential for structurally diverse VOCs to undergo unexpected autoxidation pathways, producing HOMs at greater yield and with higher molecular complexity than previously anticipated.
1 Introduction
Volatile organic compounds (VOCs) are emitted to the atmosphere in large quantities by both anthropogenic and biogenic sources and play an important role in the chemical composition of Earth's troposphere.1 The gas-phase oxidation of VOCs, as the primary chemical transformation pathway of VOCs in the atmosphere, alters their chemical properties, as well as their impact on air quality, human health, and global climate.2 The oxidation process is usually initiated by VOC reactions with strong oxidants (most importantly OH radicals but also O3 and NO3 radicals) that leads to formation of alkyl radicals (R), followed by the rapid addition of oxygen (O2) yielding peroxy radicals (RO2). The subsequent chemistry of RO2, which governs the chemical fate of the VOC, is diverse, ranging from unimolecular isomerization reactions to bimolecular reactions with other radicals such as NO, HO2, and other RO2.3,4 The molecular structure of the RO2 and the atmospheric conditions determine which reaction pathways will be competitive.5,6 The pathways that lead to formation of low-volatility organic species, as potential precursors to formation of secondary organic aerosol (SOA), a major component of atmospheric fine particulate matter, are particularly of interest to atmospheric scientists. For example, the terminating channel of RO2 bimolecular reactions, which add an oxygen-containing functional group, such as a nitrate (–ONO2) or hydroperoxy (–OOH) group, to the carbon backbone of VOC, are considered an important pathway that lowers the volatility of organic species in the atmosphere.7,8Gas-phase autoxidation, which is initiated by a RO2 unimolecular reaction pathway, is also capable of producing low-volatility organic products (Fig. 1).9,10 The key step in atmospheric autoxidation is an intramolecular RO2 hydrogen shift (H-shift) reaction, where the peroxy group abstracts an H atom from the carbon backbone of the radical to produce a hydroperoxyalkyl radical (often denoted as QOOH). An O2 molecule then quickly adds to the QOOH to form a more oxidized hydroperoxy RO2.11 These second-generation RO2 can undergo subsequent rounds of isomerization and O2 addition, leading to rapid formation of highly oxygenated oxidation products, potentially with multiple hydroperoxide groups and very low volatility. These highly oxygenated organic molecules (HOMs) have been shown to play a role in the formation of new particles and readily undergo gas-particle transfer to existing aerosol.10,12
 | ||
| Fig. 1 Key steps in autoxidation of an example VOC. The compound C1 first reacts with OH radical and O2 to form a peroxy radical (C2), which undergoes an H-shift forming a hydroperoxide functionality and a radical center on the carbon from which the H atom was abstracted, also known as a hydroperoxyalkyl radical or QOOH (C3). Subsequent addition of O2 forms a hydroperoxy RO2 (C4), which can undergo further H-shifts. The reaction sequence can either be terminated by abstracting the H atom α to the hydroperoxide group and losing a OH radical to form a ketohydroperoxide (C5), or continue by abstracting a H atom from an unsubstituted carbon to form a new, more oxidized QOOH (C6). | ||
With the recent successful reduction in urban NOx emissions, a major contributor of RO2 bimolecular loss, the formation of HOMs through RO2 autoxidation is becoming increasingly important in urban atmospheres. In regions where NO levels become sufficiently low (0.05 to 1 ppb), a condition common in nowadays North America and Europe,13–15 RO2 can survive for 10–100 s before reacting with other bimolecular reactants (Table 1), allowing RO2 H-shift chemistry even with rate coefficients as small as ∼0.05 s−1 to compete with RO2 bimolecular pathways.11 Evaluating the H-shift rate coefficients for RO2 from different classes of VOCs thus becomes essential for accurately assessing their atmospheric oxidation potential and addressing current uncertainties in SOA production.
| [NO] | k RO2+NO[NO] | τ RO2+NO |
|---|---|---|
| 0.05–0.5 ppb | 0.01–0.10 s−1 | 10–100 s |
| [HO2] | k RO2+HO2[HO2] | τ RO2+HO2 |
|---|---|---|
| 20 ppt | 0.008 s−1 | 125.0 s |
| [RO2] | k RO2+RO2[RO2] | τ RO2+RO2 |
|---|---|---|
| 20 ppt | 8 × 10−4 s−1 | 1250 s |
Previous studies have identified various general trends in H-shift reactivity that provide qualitative information about the potential for different RO2 H-shifts. Studies have shown that the rate of H-shift process is affected by the molecular structure of the involved RO2.9,18,19 At ambient temperature, the H-shift rate coefficients of RO2 from straight-chain alkanes are found to be negligible (<10−3 s−1), while certain functional groups at the carbon center from which the hydrogen is abstracted increase the lability of C–H bonds and thereby lower the barrier to H atom transfer.18,20 For example, in our recent studies on simple ether and glycol ether compounds, we demonstrate that H-shifts where the abstraction site is α to a hydroxyl group (–OH) or ether oxygen (–COC–) can proceed at rate of ∼0.1 s−1 at laboratory temperature of 294 K.21–23 H-shift reactions that involve abstraction of an aldehydic (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or allylic (–C–CC) hydrogen can have reaction rate coefficients reaching ∼1 s−1 due to stabilization of the transition state by resonance.18,19,24,25 Another factor that affects the H-shift rates is the geometry of the ring-like H-shift transition state (TS).12,18,19,26 One important feature is the number of atoms in the ring-like structure of the TS. In general, a 1,5 and 1,6 H-shift, with a TS of six and seven-membered ring, is favorable for RO2 H-shift, while TS with a smaller ring (e.g. 1,4 H-shift) is restricted due to high barriers, and formation of a larger ring (e.g. 1,8 H-shift and beyond) has been considered entropically unfavorable.12,18,19 Thus, both short- and long-range H-shifts are regarded kinetically uncompetitive and often excluded in evaluations of RO2 autoxidation chemistry.
O) or allylic (–C–CC) hydrogen can have reaction rate coefficients reaching ∼1 s−1 due to stabilization of the transition state by resonance.18,19,24,25 Another factor that affects the H-shift rates is the geometry of the ring-like H-shift transition state (TS).12,18,19,26 One important feature is the number of atoms in the ring-like structure of the TS. In general, a 1,5 and 1,6 H-shift, with a TS of six and seven-membered ring, is favorable for RO2 H-shift, while TS with a smaller ring (e.g. 1,4 H-shift) is restricted due to high barriers, and formation of a larger ring (e.g. 1,8 H-shift and beyond) has been considered entropically unfavorable.12,18,19 Thus, both short- and long-range H-shifts are regarded kinetically uncompetitive and often excluded in evaluations of RO2 autoxidation chemistry.
In this study, we report the discovery of an unexpectedly fast 1,8 H-shift reaction at ∼1 s−1 in a RO2 derived from a long-chain diether compound: 1,2-diethoxyethane (1,2-DEE, also known as ethylene glycol diethyl ether). The compound is chosen as a model system to investigate the autoxidation chemistry of long-chain ethers and glycol ethers with multiple ether linkages – a structural motif common in compounds used as organic solvents in industrial and consumer products.27–29
The general reaction mechanism for 1,2-DEE oxidation is shown in Fig. 2. To isolate the 2-RO2 1,8 H-shift from other H-shift pathways, we synthesize a selectively deuterated 1,2-DEE (1,2-DEE-d4 in Fig. 2), where hydrogen atoms on the interior carbon centers are replaced by deuterium (D) atoms. Due to changes in zero-point vibrational energy and the much lower quantum tunneling of deuterium compared to hydrogen,30–32 the fraction of 2-RO2 among all initial deuterated RO2 is enhanced, and the rates of 1,5 and 1,6 H-shifts to this peroxy radical are expected to decrease by more than an order of magnitude.11,33 Thus, in 1,2-DEE-d4, most autoxidation products from the deuterated precursor arise only from the 1,8 H-shift.
 | ||
| Fig. 2 A simplified mechanism of autoxidation chemistry of 1,2-DEE and the deuterated 1,2-DEE-d4. The branching fractions of the initial steps are denoted in purple (derived in Section S8.1). A suite of hydroperoxy RO2 can be produced from various H-shift reactions (denoted in blue) of simple RO2. Here we exclude the heteroatoms when numbering the carbons on the main chain of the compound. The H-shift rate coefficients are calculated by MC-TST method at 294 K. Production of certain RO2 from 1,2-DEE-d4 are suppressed due to kinetic isotope effect. Those pathways are not shown here. The competing RO2 + HO2 reactions and the resulting hydroperoxides are also shown. The molecular formula and molecular masses of closed-shell products are shown in green. Detailed oxidation mechanisms of 1,2-DEE and 1,2-DEE-d4 are in Section S1. | ||
To estimate the rate coefficients of major H-shift reactions in 1,2-DEE, we perform a series of chamber experiments on OH-initiated photo-oxidation of 1,2-DEE where competition between RO2 bimolecular and unimolecular chemistry are modulated by varying the HO2 concentrations, and the experimental results are analyzed with help from theoretical calculations and box model simulations. We demonstrate that the 1,8 H-shift outcompetes the 1,5 and 1,6 shifts in 1,2-DEE RO2, challenging our expectation of how autoxidation proceeds in the atmospheric degradation of ethers and likely other compounds containing heteroatoms on their chain.
2 Methods
2.1 Experimental details
The photo-oxidation reactions of 1,2-DEE (>98%, TCI) and 1,2-DEE-d4 by OH radicals are investigated in a ∼0.7 m3 Teflon environmental chamber. All experiments are performed at laboratory temperature of 294 ± 1 K and pressure of ∼993 hPa. OH radicals are produced by photolysis of H2O2 (30% by mass, Macron Fine Chemicals), which is initiated by illumination of 350 nm (Sylvania 350 Blacklight) or 254 nm (Sankyo Denki G40T10) UV lamps housed within the chamber enclosure: | (R1) |
HO2 radicals are the primary bimolecular reactant in our experiments. To study the H-shift rate coefficients of RO2 in the system, we vary the HO2 concentration across experiments to modulate the competition between bimolecular and autoxidation chemistry. HO2 is mainly produced through reaction between CH3OH (Sigma Aldrich, >99%) and OH radical:
 | (R2) |
The HO2 concentrations in the chamber are varied by changing the number and wavelength of UV lamps in the chamber enclosure to modulate OH production rates from reaction (R1), which in turn governs HO2 formation rates through reaction (R2). The 254 nm UV lights, which provide a high H2O2 photolysis rate, are used for experiments with high [HO2] where RO2 + HO2 bimolecular reactions are dominant, while the 350 nm UV lights produce low [HO2] where autoxidation chemistry becomes favorable. RO2 self-reactions (RO2 + RO2) are suppressed by ensuring that the concentrations of HO2 is much higher than those of RO2, which is achieved by using much larger concentrations of CH3OH than the VOCs so that most of OH reacts with CH3OH in the experiments. A detailed list of experimental conditions is provided in Table S1. With these conditions, the concentrations of HO2 in our experiments are estimated by box model simulation (described later) to vary from ∼1.7 × 109 molecules cm−3 (∼0.07 ppb) with one 350 nm UV light, to ∼5.8 × 1010 molecules cm−3 (∼2.4 ppb) with eight 254 nm UV lights, and are kept ∼20 times higher than total [RO2] throughout the experiments.
The reaction products are measured by a high-resolution time-of-flight chemical ionization mass spectrometer combined with low-pressure gas chromatography (GC-CIMS). Details regarding the instrument has been described elsewhere,34 and only a summary is provided here. The instrument uses CF3O− (m/z = 85) as the reagent ion, which is sensitive to multifunctional organic compounds such as hydroperoxides and nitrates.35 During GC operation, the analytes are first cryotrapped at the head of the 2 m column (Restek RTX-1701) and then separated and elute as the column temperature increases. The sample flow is diluted with N2 and transferred to the CIMS where it passes through a fluorocopolymer-coated ion-molecule reaction zone. Product ions that result from ion-molecule clustering reactions between the analytes and CF3O− ions are detected at m/z = mass neutral + 85 amu, and those resulting from fluoride transfer reactions are detected at m/z = mass neutral + 19 amu.35 In addition to forming ion clusters, certain compounds generate characteristic fragmentation ions, such as hydroperoxides producing a small fraction (a few %) of ions at m/z 63 (FCO−2), which assists in our identification of those analytes. Since we lack authentic standards for the oxidation products, the CIMS sensitivities of most analytes are estimated based on their ion-molecule collision rates with CF3O−,35 which are calculated following the method by Su and Chesnavich36 using dipole moments and polarizabilities calculated ab initio.37 The resulting estimated sensitivities, along with more details of the CIMS calibration process, are described in Section S4. Secondary losses of reaction products in the sampling system are also accounted for when quantifying the product yields, which is discussed in Section S7.
2.2 Synthesis
We synthesized via an SN2 reaction in Fig. 3 the selectively deuterated 1,2-DEE-d4 using deuterated ethylene glycol (ethylene glycol-d4) and excess ethyl iodide (EtI). Reagents were purchased from commercial sources and used as received. Synthetic details are provided in Section S2. The identity of the compound is confirmed by GC-MS analysis (Hewlett Packard 5972 with INNOWAX column). | ||
| Fig. 3 Synthetic route to 1,2-DEE-d4. | ||
2.3 Computational details
We calculate the theoretical H-shift rate coefficients of key 1,2-DEE RO2 by multi-conformer transition state theory (MC-TST),38,39 using the approach detailed elsewhere.40,41 The theoretical routine is briefly discussed. First, conformational isomers of reactants, transition states and products are sampled with the Merck Molecular Force Field method (MMFF94)42 implemented in Spartan'24 (Wavefunction, Inc.).43 Each dihedral angle is rotated 3 times (120°) for the initial sampling. The keyword “FFHINT = X∼∼+0” was used with open-shell species, to assign a charge of 0 to the radical center X. Once conformer lists were obtained, all structures were optimized at the B3LYP/6-31+G(d) level of theory44–48 with the Gaussian'16 software (Gaussian, Inc.).49 Unique conformations, filtered based on an electronic energy and electric dipole moment similarity threshold (10−5 hartree and 0.015 debye respectively), with zero-point corrected energies within 2 kcal mol−1 of the global minimum were re-optimized at the ωB97X-D/aug-cc-pVTZ level of theory.50–52 Calculations done under the Rigid-Rotor Harmonic-Oscillator (RRHO) approximation provided zero-point vibrational energies and partition functions, as well as served to ensure that each optimized structure corresponds to an appropriate potential energy surface stationary point. Intrinsic reaction coordinate (IRC) calculations, followed by geometry optimization of end-points, were done to the lowest conformer of each transition state to verify whether it is connected to the assumed reactants and products. Finally, single-point calculations at the RO-CCSD(T)-F12a/cc-pVDZ-F12 (gem_beta = 0.9)53,54 were done with Molpro 2025 (ref. 55 and 56) to the lowest conformer of each stationary point and each conformer obtained from IRC calculations, to obtain more accurate electronic energies. The RO-CCSD(T)-F12a/cc-pVDZ-F12 level-of-theory is hereinafter abbreviated as F12.Once the reaction pathway energetics were obtained, thermal rate coefficients k(T) were calculated with eqn (1):
 | (1) |
 | ||
| Fig. 4 Mechanism of 1,2-DEE 2-RO2 bimolecular reaction with HO2 (ref. 3, 58 and 59) and H-shift reactions. The H-shift rate coefficients are calculated by MC-TST method at 294 K and denoted in blue. Subsequent bimolecular reaction of 2-OO-5-ROOH + HO2 and products are also shown. Further reactions from 4-OO-2-ROOH and 3-OO-2-ROOH are shown in Schemes S2 and S3. The molecular formula and molecular masses of major oxidation products are denoted in green. Products that are detected by our instrument are shown in orange boxes. More detailed oxidation mechanisms of 2-RO2 and deuterated 2-RO2-d4 are in Section S1. | ||
To understand the observed selectivity trend in terms of molecular structures, we performed an additional set of calculations to assess the impact of inductive, stereoelectronic, and steric effects on reaction energy barriers. Inductive effects were probed with a local electrophilicity index ω+,60 whose calculation is based on the global electrophilicity index ω as defined by Parr et al.61 and the Fukui function for nucleophilic attack f+(X)62,63 condensed onto the radical-center X:
| ω+ = ωf+(X) | (2) |
 | (3) |
Steric effects were probed with Natural Steric Analysis,69,70 done with NBO7.0, and by analysis of non-covalent interactions (NCI) based on reduced density gradient plots71 and electron density (ρ) integrals over NCI domains,72 done with the Multiwfn 3.8 software.73,74 The quantitative NCI analysis employed here is based on the qbind index as a measure of interaction strength,72 where a positive or negative value indicate an overall repulsive or attractive interaction respectively, and is calculated as:
 | (4) |
2.4 Box model
We use a MATLAB-based gas-phase chemistry box model to simulate the experimental results and evaluate the kinetics and branching ratios of key oxidation reactions. The model uses the MATLAB ode15s solver to calculate time-dependent concentrations of chemical species based on given initial conditions and a specified chemical mechanism. The reactions and kinetic data implemented in the model are initially adopted from the Master Chemical Mechanism (MCM),75 estimations from literature,4,76 and calculated RO2 H-shift rate coefficients. We minimize the difference between the experimental and model results by adjusting reaction parameters of key reactions in the mechanism, including the RO2 H-shift rate coefficients and branching fractions of bimolecular reactions. Details regarding the oxidation mechanism and reaction kinetics implemented in the model is provided in Section S6.3 Results and discussion
3.1 Experimental results
Shown in Fig. 5 are the gas chromatograms of major reaction products from a 1,2-DEE and a 1,2-DEE-d4 oxidation experiment under similar conditions at HO2 level of ∼1 ppb. The deuterated oxidation products elute slightly earlier than the non-deuterated products.77 As mentioned above, we observe 2-ROOH-d4 and deuterated oxo-ROOH and DiROOH from 1,8 H-shift reaction of 2-RO2-d4 at 4 m/z units higher than their non-deuterated counterparts (e.g. m/z 239 vs. m/z 235 for 2-ROOH). Other oxidation products that result from abstraction of 1 or 2 D atoms between the ether oxygens, however, appear at only 3 or 2 m/z units higher, such as the deuterated 3-ROOH (3-ROOH-d3 at m/z 238 in Fig. 5b and Scheme S1). More detailed chromatographic analysis of 1,2-DEE oxidation products can be found in Section S1.1. From these chromatograms, we can distinguish the 1,2-DEE autoxidation products from the 1,8 H-shift reaction of 2-RO2, including 2-oxo-5-ROOH, 2-OOH-5-R′CHO, and 2,5-DiROOH, from those formed via other H-shift pathways (discussed in Section S1.1). Detection of these products at high yields confirms the occurrence of the 1,8 H-shift, and their higher yields relative to products from other autoxidation pathways suggests that the reaction outcompetes other H-shift pathways in the system.
 | ||
| Fig. 5 Gas chromatograms of major reaction products from 1,2-DEE and 1,2-DEE-d4 oxidation and tentative peak assignments. (a) 1,2-DEE oxidation products, (b) 1,2-DEE-d4 oxidation products. Certain signals are scaled up by a factor denoted in the figure. Most reaction products are detected at m/z = mass neutral + 85, except the DiROOHs which are detected at m/z = mass neutral + 85 and +19, and as other fragment ions (not shown here, see Fig. S5). Tentative assignment of different stereoisomers of DiROOHs are also denoted (discussed in Section S1.1). In this work, we refer to the stereoisomer with different configurations at both chiral centers, i.e. the meso form (R,S) and (S,R), as (R,S), and those with same configuration, i.e. (R,R) and (S,S), as (R,R). | ||
3.1.2.1 2-RO2 1,8 H-shift. The rate coefficient of the 1,8 H-shift reaction in 2-RO2 is estimated by comparing the yields of its major product from the first-generation bimolecular reaction, 2-ROOH, with the sum of the yields of autoxidation products formed following the 1,8 H-shift, including 2-oxo-5-ROOH, 2-OOH-5-R′CHO, and 2,5-DiROOH.
In absence of secondary chemistry, we assume that the ratio between the product yields is equivalent to the ratio between their production rates. By assuming steady state for the radical species, we can formulate the production rates of those compounds as:
| P2-ROOH = α2-ROOHk2-RO2+HO2[2-RO2][HO2] | (5) |
 | (6) |
 | (7) |
Consistent with eqn (7), we observe a linear relationship between the ratio of product yields and estimated [HO2] (Fig. 6). The 1,8 H-shift rate coefficient of 2-RO2 can thus be derived from the slope of the linear relationship by:
 | (8) |
 | ||
| Fig. 6 [2-ROOH]/([2-oxo-5-ROOH] + [2-OOH-5-R′CHO] + [2,5-DiROOH]) vs. [HO2] at ambient temperature (294 K), and the result from linear regression fit (R2 = 0.997). [HO2] is calculated using the box model described in Section 2.4. | ||
From linear regression fitting, we find the slope in Fig. 6 to be (1.32 ± 0.43) × 10−11 cm3 molecule−1. The rate coefficient k2-RO2+HO2 can be estimated based on method in Jenkin et al.4 and Wennberg et al.78 to be 1.96 × 10−11 cm3 molecule−1 s−1 at 294 K. We estimate the branching ratio α2-ROOH by comparing the concentrations of simple hydroperoxides (ROOH) at high [HO2], where their yields are less affected by autoxidation chemistry and are mainly controlled by the branching fractions of respective simple RO2 (αRO2) during the initiation step (Fig. 2) and the branching fractions to the formation of ROOH (αROOH) from bimolecular reaction RO2 + HO2. We estimate those parameters using box model simulations, and more details about this analysis is provided in Section S8.1.
Based on our evaluations of relevant reaction parameters listed in Table 2, we estimate the 1,8 H-shift rate coefficient of 2-RO2 to be 1.17 ± 0.85 s−1. The estimated H-shift rate coefficient agrees well with that calculated using the computational method (0.55 s−1). The source of uncertainties in the derived parameters is discussed below. We also estimate the rate coefficients of other 2-RO2 and 3-RO2 H-shift reactions (1,5 and 1,6 H-shifts) using similar measure based on the yields of other autoxidation products. More details of the estimation process are provided in Section S8.3. The average of the experimental rate coefficients of 2-RO2 and 3-RO2 1,6 H-shift reactions are estimated to be ∼0.18 s−1, and the average of those of the 1,5 H-shifts are estimated to be ∼0.06 s−1. These values also agree well with theoretical predictions, with calculated 1,6 H-shift rate coefficients of 0.11 and 0.12 s−1 for 2-RO2 and 3-RO2, respectively, and 1,5 H-shift rate coefficients of 0.02 and 0.08 s−1 (Table 4). In contrast to intuition, the entropically unfavorable 1,8 H-shift proceeds much faster than the 1,5 and 1,6 H-shifts. The rates of 1,5 and 1,6 H-shift reactions of simple 1,2-DEE RO2's are similar to those of 1,5 and 1,6 H-shifts in other organic substrates adjacent to an oxygen-containing functional group, such as hydroxy, hydroperoxy, and ether groups (generally at ∼0.05–0.2 s−1).11,20,22,23 These H-shifts are facilitated by the electron-donating α-oxygen atoms, which stabilize the H-shift TSs.25,79 The rate of the 1,8 H-shift is notably higher, approaching values reported for fast H-shifts in aldehyde, isoprene, and terpene-derived RO2.18,78,80–82 The high 2-RO2 1,8 H-shift rate coefficient likely results from a combination of factors, which are further discussed in the theoretical Section 3.2.
3.1.2.2 2-OO-5-ROOH 1,8 H-shift. The rate coefficient of the subsequent 1,8 H-shift reaction of the hydroperoxy RO2 2-OO-5-ROOH can be estimated by analyzing the variations in the yields of two of its major reaction products, 2-oxo-5-ROOH and 2,5-DiROOH (Fig. 4), as a function of [HO2]. The ratio between the yields of the two compounds, which is assumed to be equal to their production rates as above, can be formulated as:
 | (9) |
Consistent with eqn (9), there is a linear relationship between the ratio of the yields of the two compounds and 1/[HO2] (Fig. 7). We derive the H-shift rate coefficient from the slope of the linear fit in Fig. 7, with knowledge of the bimolecular rate coefficient (k2-OO-5-ROOH+HO2) and the branching ratio to formation of the dihydroperoxide (α2,5-DiROOH):
| k1,8 H-shift,2-OO-5-ROOH = α2,5-DiROOHk2-OO-5-ROOH+HO2 (slope in Fig. 7) | (10) |
 | ||
| Fig. 7 [2-oxo-5-ROOH]/[2,5-DiROOH] vs. 1/[HO2] at ambient temperature (294 K), and the result from linear regression fit (R2 = 0.979). | ||
The slope of the linear relationship is (1.22 ± 0.40) × 109 molecule cm−3. As above, the bimolecular rate coefficient is estimated using the method in Jenkin et al.4 and Wennberg et al.78 to be 2.10 × 10−11 cm3 molecule−1 s−1 at 294 K. The branching fractions of the three channels of 2-OO-5-ROOH + HO2 reaction are derived from: (1) the intercept of the linear fit, which equals to the ratio between the branching fractions of reaction channels to formation of 2-oxo-5-ROOH and 2,5-DiROOH, and (2) the yields of 2-OOH-5-R′CHO (Fig. 4), which is formed from the reaction channel to formation of alkoxy radical 2-O-5-ROOH (α2-O-5-ROOH). The resulting reaction parameters are in Table 3, with more details regarding this derivation provided in Section S8.2.
| α 2,5-DiROOH | α 2-oxo-5-ROOH | 1,8 H-shift rate coefficient (s−1) at 294 K | |
|---|---|---|---|
| Theoryb | Experiment | ||
| a Branching fractions for reaction channels of 2-OO-5-ROOH + HO2 reaction. b Calculated by MC-TST method with an uncertainty about a factor of 5. | |||
| 0.65 ± 0.09 | 0.26 ± 0.07 | 0.0038 (R,R) 8.1 × 10−4 (R,S) | 0.017 ± 0.011 (average) 0.027 ± 0.017 (R,R) 0.007 ± 0.004 (R,S) |
With these constraints, the 1,8 H-shift rate coefficient of 2-OO-5-ROOH is estimated to be 0.017 ± 0.011 s−1. We estimate stereoisomer-specific rate coefficients of this H-shift by comparing the yields of (R,S) and (R,R)-2,5-DiROOH (Fig. 5) as a function of HO2 concentrations (detailed in Section S8.2). The 1,8 H-shift rate coefficient of (R,R)-2-OO-5-ROOH is estimated to be 0.027 ± 0.017 s−1, and that of the (R,S) isomer to be 0.007 ± 0.004 s−1, which agree less closely with theoretical predictions (0.0038 s−1 and 8.1 × 10−4 s−1 for (R,R) and (R,S) stereoisomers respectively). This second-generation 1,8 H-shift is estimated to be much slower than the first-generation 1,8 H-shift process, despite the presence of an α-hydroperoxyl substituent, which should be C–H bond activating in hydrocarbon-derived systems.9,18 Similar behavior is also reported in previous studies, where α-hydroperoxyl substitution to an abstraction site that is already α-oxyl substituted decreases the RO2 H-shift rate coefficient.23,83 Theoretical evaluation of this phenomenon is also provided in Section 3.2.
3.2 Computational results
H-shift reaction energy barriers and rate coefficients calculated for major first-generation peroxy radicals, 2-RO2 and 3-RO2, and the major second generation peroxy radical, 2-OO-5-ROOH, are shown in Table 4.| Reactant | Reaction | Δ‡E0 [kcal mol−1] | k MC [s−1] | Q TS/R | κ |
|---|---|---|---|---|---|
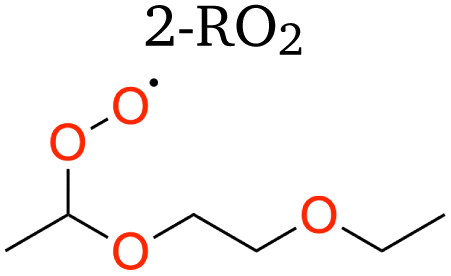
|
1,5 H-shift | 20.50 | 2.3 × 10−2 | 0.0949 | 67.4 |
| 1,6 H-shift | 18.76 | 1.1 × 10−1 | 0.0226 | 71.5 | |
| 1,8 H-shift | 16.85 | 5.5 × 10−1 | 0.0054 | 56.1 | |
|
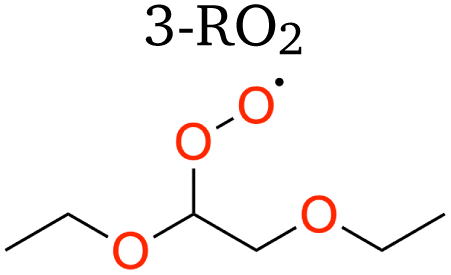
1,5 H-shift | 19.87 | 7.8 × 10−2 | 0.1130 | 66.1 |
| 1,6 H-shift (α-OR) | 18.55 | 1.2 × 10−1 | 0.0130 | 88.6 | |

|
(R,R) 1,5 H-shift (δ-OOH) | 21.03 | 5.1 × 10−3 | 0.0959 | 37.1 |
| (R,R) 1,6 H-shift (γ-OOH) | 20.30 | 1.1 × 10−2 | 0.0565 | 38.4 | |
| (R,R) 1,8 H-shift (α-OOH) | 20.09 | 3.8 × 10−3 | 0.0173 | 30.7 | |
| (R,S) 1,5 H-shift (δ-OOH) | 20.57 | 1.1 × 10−2 | 0.0523 | 63.6 | |
| (R,S) 1,6 H-shift (γ-OOH) | 19.49 | 4.8 × 10−2 | 0.0283 | 84.4 | |
| (R,S) 1,8 H-shift (α-OOH) | 20.80 | 8.1 × 10−4 | 0.0118 | 32.3 |
Consistent with the experimental findings, the ab initio calculations indicate that the 1,8 H-shift reaction in 1,2-DEE 2-RO2 is faster than the competing shorter-span pathways, 1,5 and 1,6 H-shifts. This is an unexpected finding given that longer-span H-shifts, such as 1,7 or 1,8 H-shifts, are typically observed to be less competitive due to the larger entropic penalty that accompanies the loss of many internal rotors during the formation of an eight- or nine-membered ring-like TS.18,19 While this entropic penalty is indeed larger for the 2-RO2 1,8 H-shift reaction than it is for its shorter-span counterparts, as seen from the smaller calculated ratio of partition functions QTS/R (third term in eqn (1)) in Table 4, its reaction energy barrier Δ‡E0 is also much lower, by about ∼2 and ∼4 kcal mol−1. It is known that π-donating substituents, such as oxyl (–OR) or amino (–NRR′) groups, can increase the lability of C–H bonds in α positions substantially,76 stabilizing H-shift TSs via hyperconjugation.25,79 However, all three abstraction sites in the investigated 2-RO2 H-shifts are both secondary and α-oxyl substituted, so that, in principle, this C–H bond activating effect is present regardless of the H-shift span. It is therefore clear that some other effect impacting the relative stability of H-shift TSs must be driving the observed selectivity.
hyperconjugation.25,79 However, all three abstraction sites in the investigated 2-RO2 H-shifts are both secondary and α-oxyl substituted, so that, in principle, this C–H bond activating effect is present regardless of the H-shift span. It is therefore clear that some other effect impacting the relative stability of H-shift TSs must be driving the observed selectivity.
Recent work done on similar reactions83,86 show that the presence of different heteroatoms in a radical substrate alters the selectivity of intramolecular H-shift reactions, such that the optimum span is different from what would be expected in a hydrocarbon-derived analogue. It is also shown that these differences are due, in part, to the inductive deactivation of C–H bonds by σ-withdrawing heteroatoms. While π-donation affects only the α position (in a saturated chain), the effect of electron withdrawal by electronegative atoms can propagate further along the σ-framework, polarizing the C–H bonds in β and γ positions.83,86,87 As a result, these polarized C–H bonds are less nucleophilic, and thus less susceptible to attack by an electrophilic radical center (such as a peroxy group). In the case of 1,2-DEE 2-RO2, the abstraction sites in the 1,5 and 1,6 H-shift reactions are β-oxyl substituted, whereas the site in the 1,8 H-shift is not. Reaction assistance provided by an α-oxyl group via hyperconjugation to the shorter-span H-shifts is then likely partly offset by induction from a β-oxyl group.
We tested this hypothesis by performing a series of computational investigations on H-shift reactions in a RO2 derived from a hydrocarbon analogue to 1,2-DEE, octane (OCT 2-RO2), and two RO2 derived from a monoether analogue, ethoxypentane (EP 2-RO2 and EP 7-RO2). Calculation results for these systems, as well as their structures, are shown in Fig. 8 and Sections S5.3–S5.6.
 | ||
| Fig. 8 (a) Reaction energy barriers Δ‡E0 calculated as the difference in F12//ωB97XD/aug-cc-pVTZ zero-point corrected energy between the lowest-conformer of the TS and reactant, (b) site-specific difference in local electrophilic index Δω+ = ω+(product) − ω+(reactant), (c) natural steric energy of activation Δ‡Esteric = Esteric(TS) − Esteric(reactant), and (d) analogous Δ‡qbind = qbind(TS) − qbind(reactant) NCI index for H-shift reactions in OCT 2-RO2 (black), 1,2-DEE 2-RO2 (red), EP 2-RO2 (green), and EP 7-RO2 (blue). Values of Δω+, Δ‡Esteric, and Δ‡qbind were calculated for lowest-conformers at the ωB97XD/aug-cc-pVTZ level. More details on these calculations are given in Sections S5.3–S5.6. | ||
Comparison of 1,5 H-shift reaction energy barriers in all four systems shown in Fig. 8a highlights the C–H bond deactivating effect of β-oxyl substitution: Δ‡E0 is ∼1 and ∼2 kcal mol−1 higher for 1,2-DEE 2-RO2 and EP 7-RO2 relative to EP 2-RO2 and OCT 2-RO2 respectively. This trend is reflected in the site-specific local electrophilic index differences Δω+, where the least negative values, which are associated with less nucleophilic abstraction sites,60,87,88 are those at β-oxyl positions (Fig. 8b). These findings agree with previous work investigating similar selectivity trends.83,86 Doing the same comparison with 1,6 H-shift reaction energy barriers, however, reveals the opposite behavior: Δ‡E0 is slightly lower (∼0.4 kcal mol−1) for 1,2-DEE 2-RO2 and EP 2-RO2 relative to EP 7-RO2 and OCT 2-RO2, respectively. Here, an effect associated with the fact that the β-heteroatom is endocyclic in the TS's ring-like structure, rather than exocyclic like in the case of 1,5 H-shifts, is possibly counteracting the effect of induction.
Previous studies89–91 have shown that cyclization reactions become more favorable when some of the methylene groups in the reactant's backbone are replaced by oxygen atoms. Experimental evidence92 also suggests that such observation is associated with a decrease in transannular strain. While smaller rings, such as three- or four-membered rings, suffer from a large degree of ring-strain associated with angle and torsional strain, larger rings such as seven- or eight-membered rings suffer from transannular strain, provoked by steric clashes between groups on opposite sides of the cycle.93 In the context of intramolecular H-shift reactions, some degree of transannular repulsion may destabilize the ring-like TS of longer-span pathways (1,7 or 1,8), as reported by Hao et al.86 Our calculations for OCT 2-RO2 H-shifts (Fig. 8a) show that energy barriers initially decrease with increasing span, consistent with a reduction in angle or torsional ring-strain in the TS, but then increase by ∼2 kcal mol−1 going from a 1,7 to a 1,8 H-shift span. An analogous increase in Δ‡E0 is not observed for the ether-derived 2-RO2. Moreover, the calculated 1,8 H-shift reaction energy barriers (Δ‡E0) are more than 3 kcal mol−1 lower for 1,2-DEE 2-RO2 and EP 2-RO2 relative to EP 7-RO2 and OCT 2-RO2 and respectively. Perhaps this indicates that replacing endocyclic methylene groups by oxygen atoms in longer-span RO2 H-shift TSs reduces transannular strain.
The steric energy of activation Δ‡Esteric from Natural Steric Analysis69,70 and the analogue index Δ‡qbind from NCI analysis72 calculated for the H-shift reactions investigated here (Fig. 8c and d) partly support the transannular strain hypothesis. For 1,8 H-shifts, both Δ‡Esteric and Δ‡qbind are the largest for OCT 2-RO2. These values are also the ones that see the largest increase when compared to the 1,6 H-shift, the optimal H-shift span from a TS ring-strain perspective. However, we observe some inconsistencies when comparing Δ‡Esteric and Δ‡qbind for other systems and H-shift spans. First, the 1,8 H-shift Δ‡qbind is larger for 1,2-DEE 2-RO2 than it is for EP 7-RO2, and the calculated Δ‡Esteric is almost the same for both systems, despite the lower Δ‡E0 calculated for the former. Second, ring-strain is unlikely to be the counteracting effect to induction in the 1,6 H-shift TS of 1,2-DEE 2-RO2 and EP 2-RO2 discussed above, given that transannular interactions are expected to be minimal in rings of this size, regardless of the system (see NCI domain plots in Section S5.5). Thus, we argue that transannular strain (or lack thereof) can partly explain the disparate reactivity observed for longer-span H-shifts in ether- and hydrocarbon-derived RO2, yet another structural factor is also playing a role. We hypothesize that this factor is a consequence of the generalized anomeric effect,94 and how the strength of this effect changes during the course of an H-shift reaction.
The generalized anomeric effect, often rationalized in terms of  hyperconjugative interactions,25,94,95 acts to enhance H-shift reaction rates in α-oxyl peroxyl radicals, such as 1,2-DEE 2-RO2 and EP 2-RO2, as seen in the evolution of spin-orbitals occupied by β-spin electrons (Fig. 9). As the hydrogen atom is transferred during the course of an H-shift reaction, the bonding πOO spin-orbital in the reactant is replaced by a non-bonding nO spin-orbital in the transition state. Since non-bonding orbitals are, in general, better electron donors than bonding orbitals, the
hyperconjugative interactions,25,94,95 acts to enhance H-shift reaction rates in α-oxyl peroxyl radicals, such as 1,2-DEE 2-RO2 and EP 2-RO2, as seen in the evolution of spin-orbitals occupied by β-spin electrons (Fig. 9). As the hydrogen atom is transferred during the course of an H-shift reaction, the bonding πOO spin-orbital in the reactant is replaced by a non-bonding nO spin-orbital in the transition state. Since non-bonding orbitals are, in general, better electron donors than bonding orbitals, the  interaction is stronger than the
interaction is stronger than the  interaction. The anomeric effect thus better stabilizes the transition state than it does for the reactant, lowering the reaction energy barrier. An analogous enhancement in hyperconjugative stabilization can also assist H-shift reactions in hydrocarbon-derived RO2, but to a lesser extent, because
interaction. The anomeric effect thus better stabilizes the transition state than it does for the reactant, lowering the reaction energy barrier. An analogous enhancement in hyperconjugative stabilization can also assist H-shift reactions in hydrocarbon-derived RO2, but to a lesser extent, because  orbitals are poor electron acceptors. Our hypothesis is supported by second-order perturbation theory analysis of NBO donor–acceptor interactions. Results from these calculations and a more detailed discussion of this effect are given in Section S5.7.
orbitals are poor electron acceptors. Our hypothesis is supported by second-order perturbation theory analysis of NBO donor–acceptor interactions. Results from these calculations and a more detailed discussion of this effect are given in Section S5.7.
 | ||
Fig. 9 Generalized anomeric effect in the mechanism of intramolecular H-shift reactions of α-oxyl peroxyl radicals. The relevant α spin-orbitals (top row) and β spin-orbitals (bottom row) are not shown to scale, and the wavefunction sign is indicated with purple/green color. Filled spin-orbitals are indicated with up or down arrows for α and β spin electrons respectively. As the system evolves along the reaction coordinate, a weak hyperconjugative  interaction between β spin-orbitals in the reactant (bottom row, left structure) is replaced by a strong interaction between β spin-orbitals in the reactant (bottom row, left structure) is replaced by a strong  interaction the transition state (bottom row, middle structure), stabilizing the latter relative to the former. interaction the transition state (bottom row, middle structure), stabilizing the latter relative to the former. | ||
In summary, our finding that the 1,8 H-shift reaction is the fastest amongst unimolecular pathways available to 1,2-DEE 2-RO2 can be explained by a combination of factors. C–H bond activation via hyperconjugative stabilization of the TS by an α-oxyl group, and a strengthened generalized anomeric effect at the TS relative to the reactant, both of which act to enhance the reactivity towards intramolecular H-shift reactions overall. Partial C–H bond deactivation at the abstraction site of competing pathways (1,5 and 1,6 H-shifts) via inductive electron withdrawal by a β-oxyl group, and a weaker degree of transannular repulsion experienced by the system at the 1,8 H-shift TS (compared to a hydrocarbon-derived RO2), act to enhance selectivity towards the 1,8 H-shift reaction in specific. We note that the calculated steric energy trends challenge the usual explanation that entropic factors alone are the cause for the typical lack of competitiveness of long-span RO2 H-shifts. While the large entropic penalty from the loss of many internal rotors is certainly important, TS energy-raising transannular repulsion is apparently also part of the reason why 1,8 H-shifts are slower in hydrocarbon-derived RO2.
Regarding the second-generation 1,8 H-shift, i.e. the 1,8 H-shift reaction of 2-OO-5-ROOH, we notice that it is much slower than the first-generation 2-RO2 1,8 H-shift (Tables 2 and 3), despite that the H abstraction site is substituted with two π-donating functional groups: –OOH and –O–.25,79 Here, C–H bond activation via π-donation from both substituents may not be entirely additive, while being overwhelmed by inductive C–H bond deactivation via σ-withdrawal by two electronegative α-oxygen atoms.87,96 Moreover, the intramolecular hydrogen-bond (H-bond) between the –OOH and –O– groups in the RO2 reactant, which is exchanged by a weaker H-bond between the –OOH and the –OO groups in the TS of H-abstraction from the α-OOH position, likely also plays a role (Section S5.8). The additional energy which is required to break this intramolecular H-bond raises the energy barrier for this H-shift reaction pathway. Presence of this H-bond also makes the 1,8 H-shift a minor reaction channel for 2-OO-5-ROOH, outcompeted by its 1,5 and 1,6 H-shift pathways (Table 4 and Section S8.2), whose reaction TS formation does not involve disrupting the intramolecular interaction.
3.3 Atmospheric implications
Based on our estimation, the fast 2-RO2 1,8 H-shift reaction can rival their reactions with NO at a NO level of ∼5 ppb at 294 K (with kRO2+NO = 9.2 × 10−12 cm3 molecule−1 s−1 based on Jenkin et al.4). Since RO2 H-shift rates generally increase with temperature due to the energy barrier associated with the H-shift reaction coordinate,3,12 we expect the H-shift reaction to be competitive with bimolecular reaction with NO at even higher NO levels under warmer conditions. As shown in Table 5, on warm summer afternoons when outdoor temperatures can exceed 30 °C and occasionally reach 40 °C, the H-shift pathway can proceed at rates greater than 2 s−1 and outrun bimolecular reactions at [NO] > 9 ppb. As urban NOx levels often fall below 1 ppb due to successful NOx emission control,14,15 the H-shift reaction is expected to be the dominant reaction pathway for the RO2 under most urban atmospheric conditions. equivalent to the H-shift rates
equivalent to the H-shift rates
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(360/
exp(360/Atmospheric autoxidation chemistry produces low-volatility HOMs through consecutive RO2 H-shift reactions and O2 addition, which add multiple hydroperoxy functionalities onto the carbon backbone of the VOC until the reaction chain is terminated by H abstraction at an α-OOH position, forming a ketone group (Fig. 1). VOCs with sufficiently long carbon chain to support multi-generational H-shifts are thus considered to have a high potential to produce di- or even trihydroperoxy oxidation products, which will have extremely low volatility and promote new particle formation.12 The long-chain diether compound investigated here represents such a system, and the fast long-range H-shift reaction further enhances its potential for HOM and SOA formation by enabling rapid incorporation of hydroperoxy groups at all possible positions in the molecule. The reaction allows the formation of HOMs from long-chain VOCs with the diether moiety, such as long-chain glycol ethers,28 including those structurally similar to 1,2-DEE,29 at a much higher rate than previously believed. For long-chain VOCs containing other oxygenated functional groups or other heteroatoms, such long-range RO2 H-shift may also be possible,83 expanding the range of viable autoxidation pathways that produce structurally complex HOMs.
4 Conclusions
We perform experimental and computational investigations on intramolecular H-shift reactions of RO2 derived from OH-initiated oxidation of the diether 1,2-DEE, and identify an unexpectedly fast 1,8 H-shift reaction of 1,2-DEE 2-RO2 occurs at ∼1 s−1 at ambient temperature of 294 K. This long-span H-shift outruns not only all other H-shift reactions in 1,2-DEE-derived RO2, but also their bimolecular chemistry under atmospheric conditions. Quantum chemical calculations attribute the exceptional rate of this long-range H-shift reaction to a combination of factors. Hyperconjugative C–H bond activation by an α-oxyl group, combined with a kinetic generalized anomeric effect involving the peroxy group, enhances the RO2 reactivity towards H-shifts overall, while weakened transannular strain in the 1,8 H-shift TS, combined with inductive C–H bond deactivation by a β-oxyl group in competing shorter-span pathways, enhances the RO2 selectivity towards the 1,8 H-shift in specific. A series of photo-oxidation experiments are conducted using both ordinary and isotope-labeled 1,2-DEE, which provides validation for the computational predictions of various H-shift processes in the system.Our findings highlight how heteroatom-induced electronic effects modulate the reactivity of RO2, and in particular, how the presence and positioning of oxygen atoms and oxygen-containing functional groups in the carbon backbone influence RO2 H-shift reactions. With the unusually fast H-shift reaction identified here, a substantial fraction of 1,2-DEE RO2 (>60%) will undergo autoxidation pathways under modern urban atmospheric conditions, where typical RO2 bimolecular lifetime exceeds 5 s,11 leading to efficient formation of HOMs. We expect that similar intramolecular H-shift mechanisms can be important, yet have been previously overlooked, in many similar systems, especially long-chain ethers and glycol ethers that are important classes of volatile chemical products (VCPs),28,97 an emerging urban VOC emission source.98,99 It is also possible that the long-span H-shift chemistry is relevant in the atmospheric oxidation of non-hydrocarbon compounds containing other heteroatoms with similar electron-directing ability as oxygen,83 such as amines (R–NH–R′) and sulfides (R–S–R′), which are emitted into the atmosphere from various sources100,101 and are shown to undergo autoxidation.21,102,103 Extending investigations to those systems are essential for constraining autoxidation mechanisms and improving predictive models of HOMs and SOA formation from diverse VOC precursors. Moreover, since autoxidation chemistry via RO2 H-shift reactions is also important in low-temperature combustion, food industry, and biochemistry,104,105 our discovery here may have implications for radical chemistry in other chemical environments.
Author contributions
Conceptualization: H. Y. and P. O. W. Experimental methodology: H. Y., J. D. C. and P. O. W. Synthetic methodology: S. P. R. and B. M. S. Computational methodology: T. G. A. and H. G. K. Box model methodology: J. D. C. Experimentation: H. Y. Synthesis: S. P. R. Computation: T. G. A. and V.-T. S. Formal analysis: H. Y. and T. G. A. Writing – original draft: H. Y. Writing – review and editing: all.Conflicts of interest
There are no conflicts to declare.Data availability
Mechanism files of 1,2-DEE photo-oxidation used in the box model are available at: https://doi.org/10.22002/h46t8-rc655. Output files from all theoretical calculations are available at: https://www.erda.dk/archives/6b6758bea7b2c71578d91661942be8e5/published-archive.html.Supplementary information (SI): additional oxidation machanisms of 1,2-DEE and 1,2-DEE-d4; details about synthetic procedures; additional computational results; details about box model implementation; experimental conditions and results; additional experimental results and discussion; estimation of uncertainties. See DOI: https://doi.org/10.1039/d5sc10150f.
Acknowledgements
This work is supported by funding from U.S. National Science Foundation (CHE-2305204), Independent Research Fund Denmark (10.46540/4286-00306B), and Carlsberg Foundation (CF24-1034). We thank Gautham Kappaganthula and Katherine Ball for support in analysis of high-resolution mass-spectrometric data, Andras Sun Poulsen for the calculation of dipole moments and polarizabilities and Jing Chen and Josefine E. Borcher for initial calculations. We also thank the High Performance Computing Center at the University of Copenhagen for providing computational resources.Notes and references
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed
.
- M. Glasius and A. H. Goldstein, Environ. Sci. Technol., 2016, 50, 2754–2764 CrossRef CAS PubMed
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS
- A. Mellouki, T. J. Wallington and J. Chen, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed
- V. P. Barber and J. H. Kroll, J. Phys. Chem. A, 2021, 125, 10264–10279 CrossRef CAS PubMed
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Inc., Hoboken, New Jersey, 3rd edn, 2016 Search PubMed
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS
- M. Ehn, T. Berndt, J. Wildt and T. Mentel, Int. J. Chem. Kinet., 2017, 49, 821–831 CrossRef CAS
- E. Praske, R. V. Otkjær, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 64–69 CrossRef CAS PubMed
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed
- A. R. Russell, L. C. Valin and R. C. Cohen, Atmos. Chem. Phys., 2012, 12, 12197–12209 CrossRef CAS
- B. N. Duncan, L. N. Lamsal, A. M. Thompson, Y. Yoshida, Z. Lu, D. G. Streets, M. M. Hurwitz and K. E. Pickering, J. Geophys. Res.: Atmos., 2016, 121, 976–996 CrossRef CAS
- A. Fortems-Cheiney, G. Broquet, I. Pison, M. Saunois, E. Potier, A. Berchet, G. Dufour, G. Siour, H. D. van der Gon, S. N. Dellaert and K. F. Boersma, Geophys. Res. Lett., 2021, 48, e2020GL092206 Search PubMed
- X. Ren, H. Harder, M. Martinez, R. L. Lesher, A. Oliger, T. Shirley, J. Adams, J. B. Simpas and W. H. Brune, Atmos. Environ., 2003, 37, 3627–3637 CrossRef CAS
- X. Ma, Z. Tan, K. Lu, X. Yang, X. Chen, H. Wang, S. Chen, X. Fang, S. Li, X. Li, J. Liu, Y. Liu, S. Lou, W. Qiu, H. Wang, L. Zeng and Y. Zhang, Atmos. Chem. Phys., 2022, 22, 7005–7028 CrossRef CAS
- R. V. Otkjær, H. H. Jakobsen, C. M. Tram and H. G. Kjaergaard, J. Phys. Chem. A, 2018, 122, 8665–8673 Search PubMed
- L. Vereecken and B. Nozière, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS
- E. Praske, R. V. Otkjær, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2019, 123, 590–600 CrossRef CAS PubMed
- K. H. Møller, T. Berndt and H. G. Kjaergaard, Environ. Sci. Technol., 2020, 54, 11087–11099 CrossRef PubMed
- H. Yu, K. H. Møller, R. S. Buenconsejo, J. D. Crounse, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2023, 127, 9564–9579 CrossRef CAS PubMed
- H. Yu, J. Chen, J. D. Crounse, T. G. Almeida, H. G. Kjaergaard and P. O. Wennberg, ACS ES&T Air, 2025, 2, 2566–2576 Search PubMed
- M. P. Rissanen, T. Kurtén, M. Sipilä, J. A. Thornton, J. Kangasluoma, N. Sarnela, H. Junninen, S. Jørgensen, S. Schallhart, M. K. Kajos, R. Taipale, M. Springer, T. F. Mentel, T. Ruuskanen, T. Petäjä, D. R. Worsnop, H. G. Kjaergaard and M. Ehn, J. Am. Chem. Soc., 2014, 136, 15596–15606 CrossRef CAS PubMed
- I. V. Alabugin, G. dos Passos Gomes and M. A. Abdo, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1389 Search PubMed
- L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2010, 12, 12608–12620 RSC
- J. Moriarty, H. Sidebottom, J. Wenger, A. Mellouki and G. L. Bras, J. Phys. Chem. A, 2003, 107, 1499–1505 CrossRef CAS
- L. Li and D. R. Cocker, Atmos. Environ., 2018, 180, 206–215 CrossRef CAS
- T. R. Melles, A. V. Lawrence, A. Ksaibati, C. L. Zang, A. Dearden, M. M. Coggon, K. L. Richard, D. Ketcherside, L. Tan, C. E. Stockwell, H. Luo, M. Akbarzadeh, L. Xu, A. M. Middlebrook, A. Piasecki, L. A. Garofalo, C. Warneke, L. Hu, D. K. Farmer, S. H. Jathar and M. D. Willis, ACS Earth Space Chem., 2025, 10, 131–147 CrossRef
- A. M. Sage and N. M. Donahue, J. Photochem. Photobiol., A, 2005, 176, 238–249 Search PubMed
- H. Muchalski, A. J. Levonyak, L. Xu, K. U. Ingold and N. A. Porter, J. Am. Chem. Soc., 2015, 137, 94–97 CrossRef CAS PubMed
- M. Meder, O. Peräkylä, J. G. Varelas, J. Luo, R. Cai, Y. Zhang, T. Kurtén, M. Riva, M. Rissanen, F. M. Geiger, R. J. Thomson and M. Ehn, Atmos. Chem. Phys., 2023, 23, 4373–4390 CrossRef CAS
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC
- K. T. Vasquez, H. M. Allen, J. D. Crounse, E. Praske, L. Xu, A. C. Noelscher and P. O. Wennberg, Atmos. Meas. Tech., 2018, 11, 6815–6832 CrossRef CAS
- J. D. Crounse, K. A. McKinney, A. J. Kwan and P. O. Wennberg, Anal. Chem., 2006, 78, 6726–6732 CrossRef CAS PubMed
- T. Su and W. J. Chesnavich, J. Chem. Phys., 1982, 76, 5183 CrossRef CAS
- A. L. Garden, F. Paulot, J. D. Crounse, I. J. Maxwell-Cameron, P. O. Wennberg and H. G. Kjaergaard, Chem. Phys. Lett., 2009, 474, 45–50 CrossRef CAS
- M. Karplus and J. N. Kushick, Macromolecules, 2002, 14, 325–332 CrossRef
- L. Vereecken and J. Peeters, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS
- K. H. Møller, R. V. Otkjær, N. Hyttinen, T. Kurtén and H. G. Kjaergaard, J. Phys. Chem. A, 2016, 120, 10072–10087 Search PubMed
- Q. Zhao, K. H. Møller, J. Chen and H. G. Kjaergaard, J. Phys. Chem. A, 2022, 126, 6483–6494 Search PubMed
- T. A. Halgren, J. Comput. Chem., 1999, 20, 730–748 CrossRef CAS PubMed
-
Spartan 24, version 1.2.0, Wavefunction Inc., Irvine, CA, 2024 Search PubMed
- R. Ditchfield, W. J. Hehre, J. A. Pople, R. Ditcrfield, W. J. Herre and D. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS
- W. J. Hehre, R. Ditchfield, J. A. Pople, J. C. Phys, F. T. Wall, L. A. Hiller, D. J. Wheeler, J. C. Phys, J. M. Hammersley, K. W. Morton, J. R. S. Soc, W. J. Herre and R. Ditcrfield, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
- T. H. Dunning and T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 Search PubMed
- K. A. Peterson, T. B. Adler and H. J. Werner, J. Chem. Phys., 2008, 128, 84102 Search PubMed
- G. Knizia, T. B. Adler and H. J. Werner, J. Chem. Phys., 2009, 130, 54104 CrossRef PubMed
- H. J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS
- C. Eckart, Phys. Rev., 1930, 35, 1303 CrossRef CAS
- G. Ghigo, A. Maranzana and G. Tonachini, J. Chem. Phys., 2003, 118, 10575–10583 CrossRef CAS
- V. T. Salo, R. Valiev, S. Lehtola and T. Kurtén, J. Phys. Chem. A, 2022, 126, 4046–4056 CrossRef CAS PubMed
- F. D. Vleeschouwer, V. V. Speybroeck, M. Waroquier, P. Geerlings and F. D. Proft, Org. Lett., 2007, 9, 2720–2724 CrossRef PubMed
- R. G. Parr, L. V. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS
- W. Yang and W. J. Mortier, J. Am. Chem. Soc., 1986, 108, 5708–5711 CrossRef CAS PubMed
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS
- J. K. Badenhoop and F. Weinhold, J. Chem. Phys., 1997, 107, 5406–5421 Search PubMed
- J. K. Badenhoop and F. Weinhold, Int. J. Quantum Chem., 1999, 72, 269–280 CrossRef CAS
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- J. Contreras-García, W. Yang and E. R. Johnson, J. Phys. Chem. A, 2011, 115, 12983–12990 CrossRef PubMed
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- T. Lu, J. Chem. Phys., 2024, 161, 82503 CrossRef CAS PubMed
- Master Chemical Mechanisms (v3.3.1), https://mcm.york.ac.uk/MCM.
- M. E. Jenkin, R. Valorso, B. Aumont, A. R. Rickard and T. J. Wallington, Atmos. Chem. Phys., 2018, 18, 9297–9328 CrossRef CAS
- N. Thakur, S. Aslani and D. W. Armstrong, Anal. Chim. Acta, 2021, 1165, 338490 CrossRef CAS PubMed
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. S. Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed
- D. J. Henry, C. J. Parkinson, P. M. Mayer and L. Radom, J. Phys. Chem. A, 2001, 105, 6750–6756 Search PubMed
- A. P. Teng, J. D. Crounse and P. O. Wennberg, J. Am. Chem. Soc., 2017, 139, 5367–5377 Search PubMed
- L. Xu, K. H. Møller, J. D. Crounse, R. V. Otkjær, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2019, 123, 1661–1674 CrossRef CAS PubMed
- S. Barua, S. Iyer, A. Kumar, P. Seal and M. Rissanen, Atmos. Chem. Phys., 2023, 23, 10517–10532 CrossRef CAS
- J. E. Borcher, V.-T. Salo, T. G. Almeida and H. G. Kjaergaard, J. Phys. Chem. A, 2026, 130, 1375–1383 CrossRef CAS PubMed
-
J. B. Burkholder, S. P. Sander, J. P. D. Abbatt, J. R. Barker, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, Jet Propulsion Laboratory, California Institute of Technology Technical Report, 2020 Search PubMed
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS
- Z. Hao, W. Ai and N. Q. Su, J. Phys. Chem. A, 2025, 129, 4969–4978 CrossRef CAS PubMed
- J. J. Garwood, A. D. Chen and D. A. Nagib, J. Am. Chem. Soc., 2024, 146, 28034–28059 CAS
- A. Velloth, P. Kumar, S. Butt and S. Venkataramani, Asian J. Org. Chem., 2025, 14, e202400686 CrossRef CAS
- L. Mandolini, Adv. Phys. Org. Chem., 1986, 22, 1–111 CrossRef CAS
- M. A. Casadei, C. Galli and L. Mandolini, J. Am. Chem. Soc., 2002, 106, 1051–1056 CrossRef
- G. Illuminati, L. Mandolini, K. Ziegler, H. Eberle, H. Ohlinger, R. Bird, A. C. Knipe, C. J. M. Stirling and J. C. Soc, Acc. Chem. Res., 2002, 14, 95–102 CrossRef
- J. Dale, Tetrahedron, 1974, 30, 1683–1694 CrossRef CAS
- M. A. Winnik, Chem. Rev., 2002, 81, 491–524 CrossRef
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil', I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent'Ev and M. A. Zolfigol, Chem. Soc. Rev., 2021, 50, 10253–10345 RSC
- S. David, O. Eisenstein, W. J. Hehre, L. Salem and H. Roald, J. Am. Chem. Soc., 1973, 95, 3806–3807 CrossRef CAS
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5(7), 486–499 CrossRef CAS PubMed
- W. Li, L. Li, C. li Chen, M. Kacarab, W. Peng, D. Price, J. Xu and D. R. Cocker, Atmos. Environ., 2018, 178, 109–117 CrossRef CAS
- B. C. McDonald, J. A. D. Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S. W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Science, 2018, 359, 760–764 CrossRef CAS PubMed
- M. M. Coggon, G. I. Gkatzelis, B. C. McDonald, J. B. Gilman, R. H. Schwantes, N. Abuhassan, K. C. Aikin, M. F. Arendd, T. A. Berkoff, S. S. Brown, T. L. Campos, R. R. Dickerson, G. Gronoff, J. F. Hurley, G. Isaacman-Vanwertz, A. R. Koss, M. Li, S. A. McKeen, F. Moshary, J. Peischl, V. Pospisilova, X. Ren, A. Wilson, Y. Wu, M. Trainer and C. Warneke, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026653118 CrossRef CAS PubMed
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 524–546 CrossRef CAS
- P. R. Veres, J. A. Neuman, T. H. Bertram, E. Assaf, G. M. Wolfe, C. J. Williamson, B. Weinzierl, S. Tilmes, C. R. Thompson, A. B. Thames, J. C. Schroder, A. Saiz-Lopez, A. W. Rollins, J. M. Roberts, D. Price, J. Peischl, B. A. Nault, K. H. Møller, D. O. Miller, S. Meinardi, Q. Li, J. F. Lamarque, A. Kupc, H. G. Kjaergaard, D. Kinnison, J. L. Jimenez, C. M. Jernigan, R. S. Hornbrook, A. Hills, M. Dollner, D. A. Day, C. A. Cuevas, P. Campuzano-Jost, J. Burkholder, T. P. Bui, W. H. Brune, S. S. Brown, C. A. Brock, I. Bourgeois, D. R. Blake, E. C. Apel and T. B. Ryerson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4505–4510 CrossRef CAS PubMed
- Q. Ye, M. B. Goss, G. Isaacman-Vanwertz, A. Zaytsev, P. Massoli, C. Lim, P. Croteau, M. Canagaratna, D. A. Knopf, F. N. Keutsch, C. L. Heald and J. H. Kroll, ACS Earth Space Chem., 2021, 5, 2013–2020 CrossRef CAS
- E. R. Kjærgaard, K. H. Møller, T. Berndt and H. G. Kjaergaard, J. Phys. Chem. A, 2023, 127, 8623–8632 Search PubMed
- R. A. Cox and J. A. Cole, Combust. Flame, 1985, 60, 109–123 CrossRef CAS
- K. U. Ingold, Chem. Rev., 1961, 61, 563–589 Search PubMed
| This journal is © The Royal Society of Chemistry 2026 |