
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–metal vs. metal–ligand cooperation in iron-mediated activation and catalytic reduction of nitrous oxide and nitrobenzene
Nadir
Jori
 a,
Helena
Corona
a,
Enrique
Soto
a,
Israel
Fernández
*b and
Jesús
Campos
*a
a,
Helena
Corona
a,
Enrique
Soto
a,
Israel
Fernández
*b and
Jesús
Campos
*a
aInstituto de Investigaciones Químicas (IIQ), Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es
bDepartamento de Química Orgánica and Centro de Innovación en Química Avanzada, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Spain. E-mail: israel@quim.ucm.es
First published on 3rd March 2026
Abstract
There is an increasing interest in developing catalysts for transforming nitrous oxide (N2O), though systems based on Earth-abundant metals are limited. We report the synthesis of [FeII(depe)(Cp*)N2][BArF] (2; depe = 1,2-bis(diethylphosphino)-ethane; Cp* = η5-C5Me5) and its intriguing reactivity with N2O, which involves a complex manifold of competing routes. This includes an exotic metal–ligand cooperation pathway towards a Cp*-tethered hydrazinyl oxide (−O–N(R)–N(H)2) moiety competing with a metal–metal cooperation route to form a transient FeIV-oxo species. The latter leads to [FeIII(depe)(Cp*)(OH)][BArF] (3) and a product with a dehydrogenated phosphine arm (5). Precursor 2 also reacts with nitrobenzene, allowing the identification of a unique nitrosobenzene complex, [FeII(depe)(Cp*)(κ1N–C6H5NO)][BArF] (6-N), via bimetallic cooperation. The proposed mechanisms are supported by experimental methods and DFT calculations. Furthermore, 2 is an efficient catalyst for the O-transfer reaction from N2O to HBpin (pinacolborane), as well as for the reduction of nitrobenzene, where it exhibits superior performance to current homogeneous catalysts.
Introduction
The considerable increase of nitrous oxide (N2O) levels in the atmosphere has raised some major concerns, as it is one of the greenhouse gases contributing to global warming and the depletion of ozone in the atmosphere.1 Therefore, a pressing need for its degradation has spanned the development of catalysts capable of its transformation to more inert products, such as dinitrogen (N2).2,3 In this context, a wide range of heterogeneous catalysts for the conversion of N2O into higher-added value chemicals has been reported.4–6In contrast, homogeneous catalysts for the transformation of N2O are comparatively far more limited.7 They rely on the deoxygenation of nitrous oxide via O-transfer reactions to different substrates, for which a variety of molecular catalysts based on precious metals has been used.2,8–17 In contrast, the development of environmentally friendly, inexpensive catalysts based on non-precious first-row metals has been a major recent focus,18,19 which is starting to permeate into N2O catalytic protocols. Although this has so far been mostly restricted to nickel,20–23 two recent reports from Chaplin and Mo reveal the potential of copper24 and iron,25 respectively, to catalytically transform N2O (Fig. 1, top). Besides, recent studies revealed that Fe-porphyrin complexes are also competent for N2O reduction processes.26,27 Alternatively, catalysts based on main group elements have recently shown great potential for N2O reduction.28–30 Nonetheless, there is a need to better understand the operating mechanisms, often difficult to investigate for these transformations, in order to speed up catalyst development.31
 | ||
| Fig. 1 Selected examples of first-row transition metal catalysts for the reduction of N2O (top) and the cooperative iron system investigated in this work (bottom). | ||
Being the most abundant transition metal in the Earth's crust, the aforesaid use of iron for N2O conversion is particularly appealing. We have recently explored the cooperative activation of the isoelectronic CO2 molecule using [Fe0(depe)2] (depe = 1,2-bis(diethylphosphino)-ethane), which however decomposes under an N2O atmosphere.32 To provide further stability while enabling the iron centre to accommodate a wide range of oxidation states, we have now focused on its derivative [FeII(depe)(Cp*)Cl] (1; Cp* = η5-C5Me5). Our choice was encouraged by the fact that the only prior iron system to mediate the catalytic reduction of N2O relied on metal–ligand cooperation.25 Moreover, the non-innocent role of the Cp* ligand in compounds like 1 is well-documented33–36 and we hypothesized that it might aid N2O activation and catalysis. In this work, we report the synthesis, characterization and reactivity studies of the iron motif [Fe(depe)(Cp*)] in various oxidation states with N2O and nitrobenzene, including a deep mechanistic understanding through experimental and computational means. In addition, we describe the catalytic reduction of these molecules with pinacol borane.
Results and discussion
Synthesis and characterization of Fe(II) complexes
We first attempted the synthesis of 1 through previously reported routes for analogous complexes with different diphosphines (dppe = 1,2-bis(diphenylphosphino)-ethane, dippe = 1,2-bis(diisopropylphosphino)-ethane or dnppe = 1,2-bis(di-n-propylphosphino)-ethane),37–39 but they led to the concomitant formation of [FeII(depe)2Cl2] and [FeII(Cp*)2], and prevented the isolation of pure complex 1 in sufficient yield. However, 1 could be synthesized by the reaction between depe and the previously reported complex [FeII(tmeda)(Cp*)Cl] (tmeda = tetramethylethylenediamine), A,40 in THF solution at −80 °C (Scheme 1). Upon addition of the phosphine ligand, the solution changes colour from light green-yellow to deep purple. Evaporation of the solvent and extraction with pentane afforded a clear dark violet-purple solution. Dark violet needles could be grown from a saturated pentane solution at −35 °C over the course of 20 h in 82% yield. Other attempted reaction conditions gave poorer yields or led to the concomitant formation of [FeII(Cp*)2] and/or [FeII(depe)2Cl2].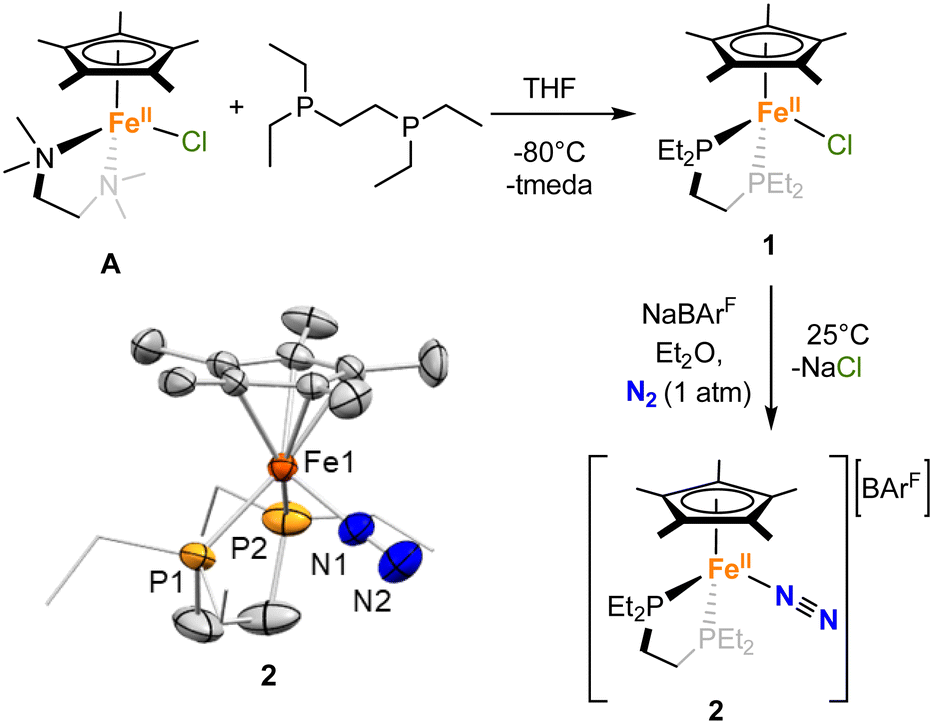 | ||
| Scheme 1 Synthesis of [Fe(depe)(Cp*)Cl] (1) and [Fe(depe)(Cp*)N2][BArF] (2). ORTEP of 2 with hydrogen atoms and the [BArF]− counterion have been excluded for clarity. Thermal ellipsoids are set at 50% probability. | ||
To activate N2O, a vacant site at the metal centre would be required. To achieve this, and as reported for previous Fe(II) half sandwich complexes,39,41,42 removal of the chloride anion from 1 under a dinitrogen atmosphere resulted in the formation of the terminal end-on N2-complex 2, [FeII(depe)(Cp*)(N2)][BArF] ([BArF]− = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate). More precisely, the addition of NaBArF to a violet-purple solution 1 in Et2O under N2 resulted in an orange suspension over the course of 3 h (Scheme 1). Upon filtration, the orange solution was stored at −35 °C, affording orange crystals of 2 over the course of 2 days in 92% yield. The formation of 2 was inferred from a slightly down-shifted 31P{1H} NMR resonance at 79.3 ppm (c.f. 83.8 for 1). Besides, the solid-state IR spectrum of 2 shows a νNN band at 2106 cm−1 (Fig. S75), which compares well with the literature values for analogous half-sandwich complexes of Fe(II) (2098–2117 cm−1),34,38,41 suggesting a low degree of activation of the N–N bond.
As expected, the solid-state molecular structures of 1 and 2 (Scheme 1 and Fig. S71) corroborate the piano-stool coordination of the Fe centers, with one leg occupied by either a chloride (1) or a N2 (2) ligand. Despite the neutral vs. ionic nature of 1 and 2, the Fe(II) center in both complexes exhibits comparable Fe–Cp* centroid (1.721(2) and 1.748(8) Å) and Fe–P (2.2133(6) and 2.2341(18) Å on average) distances. The Fe–Cl bond distance in 1 (2.3441(6) Å) falls within the range of previously reported half-sandwich Fe(II) complexes bearing Cp* and phosphine ligands (2.342–2.34(1) Å).39,40,43 Similarly, the Fe–N distance in 2 (1.847(6) Å), together with the very short distance observed between the two nitrogen atoms (0.828(12) Å) falls within the range of what was reported previously and support the formulation of 2 as an Fe(II) complex bearing a N2 neutral ligand with little activation of the N–N bond, as confirmed by IR spectroscopy (vide supra).39,42
Stoichiometric reactivity with N2O and nitrobenzene
With 2 in hand, we explored its reactivity with N2O (Scheme 2). Addition of 1 bar of N2O to a light orange-yellow THF solution of 2 at 25 °C led to slow consumption of the starting material over the course of 24 h, as confirmed by multinuclear NMR studies and also a change of color to dark orange-red (Fig. S7–S9). However, the formation of paramagnetic species over the course of the reaction prevents further characterization by NMR spectroscopy. The reaction in Et2O solution or in a toluene suspension yielded similar results (see the SI).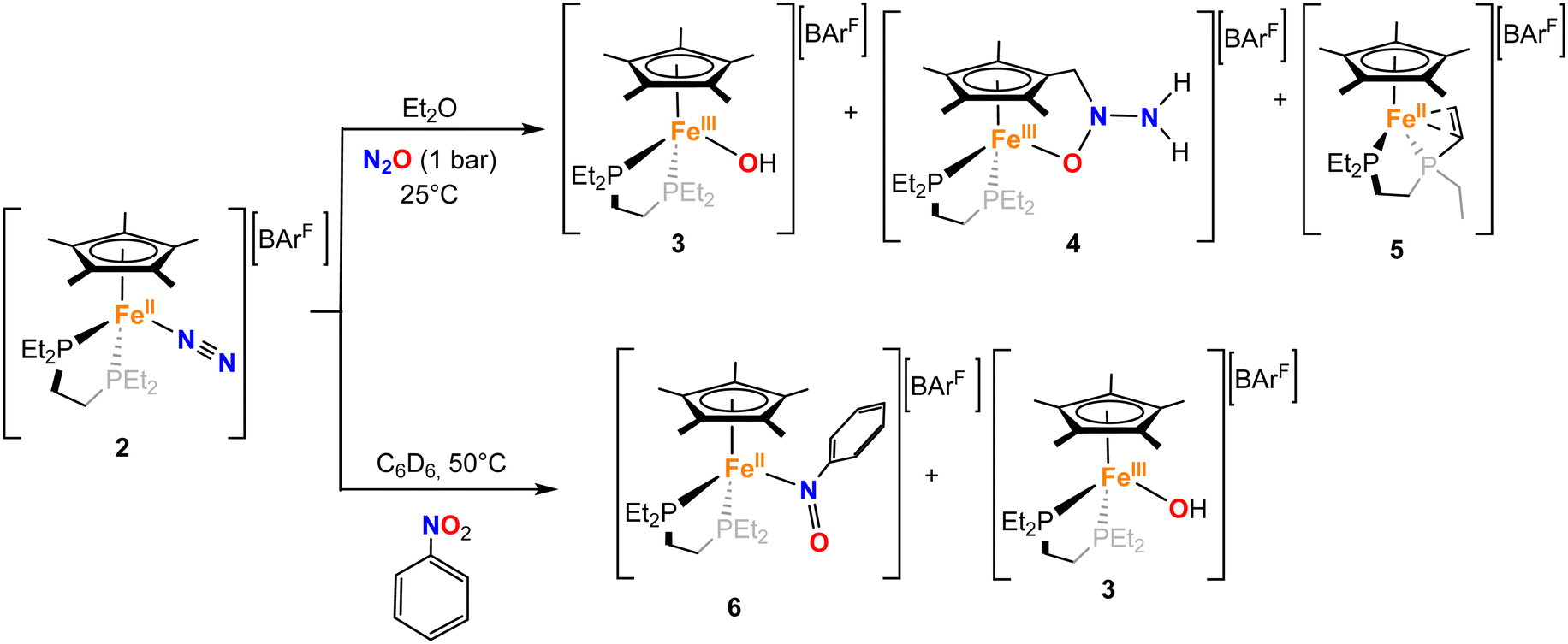 | ||
| Scheme 2 Reactivity of [Fe(depe)(Cp*)(N2)][BArF] (2) with N2O (top) and nitrobenzene (bottom). | ||
X-band EPR studies of the reaction mixture at 77 K indicate the presence of multiple Fe(III) paramagnetic species (Fig. 2). Indeed, we managed to grow crystals of three different species from vapor diffusion of pentane into an Et2O solution of the reaction mixture at −35 °C over the course of 2 days (Scheme 2 and Fig. 3).
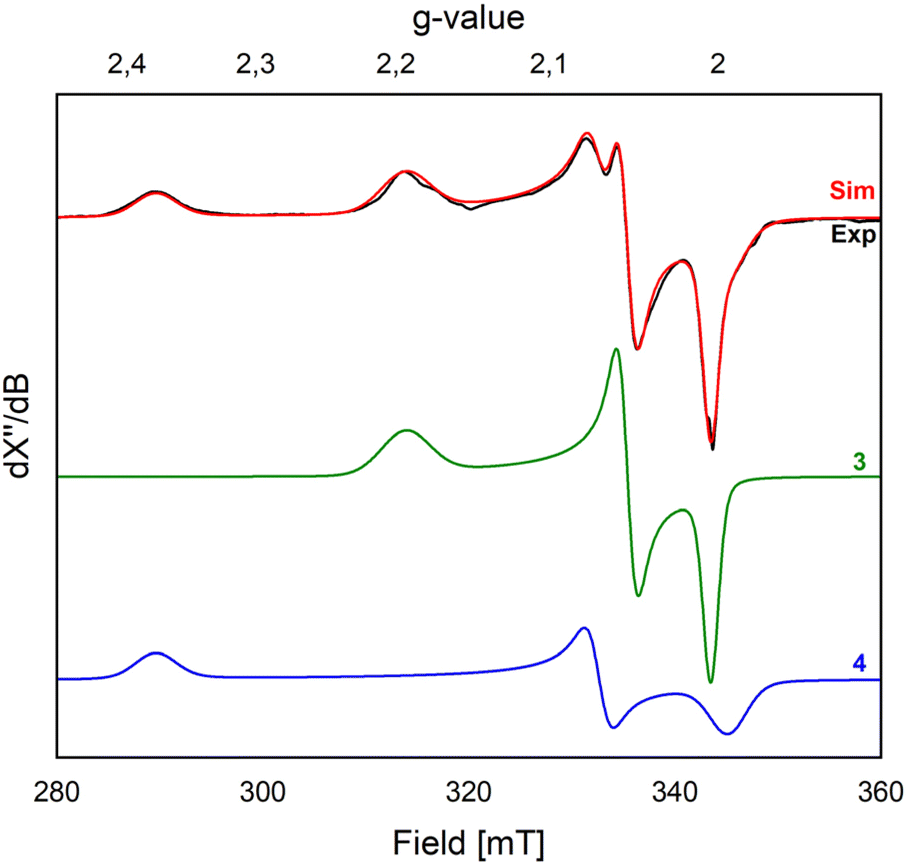 | ||
Fig. 2 EPR spectra of a frozen solution (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O = 1:1) of crystals isolated from the reaction of 2 with N2O yielding 3 and 4 (modulation amplitude: 4 G, 9.635 GHz, 77 K, 4 scans; each individual simulation (3 and 4) is weighted at 50%). :Et2O = 1:1) of crystals isolated from the reaction of 2 with N2O yielding 3 and 4 (modulation amplitude: 4 G, 9.635 GHz, 77 K, 4 scans; each individual simulation (3 and 4) is weighted at 50%). | ||
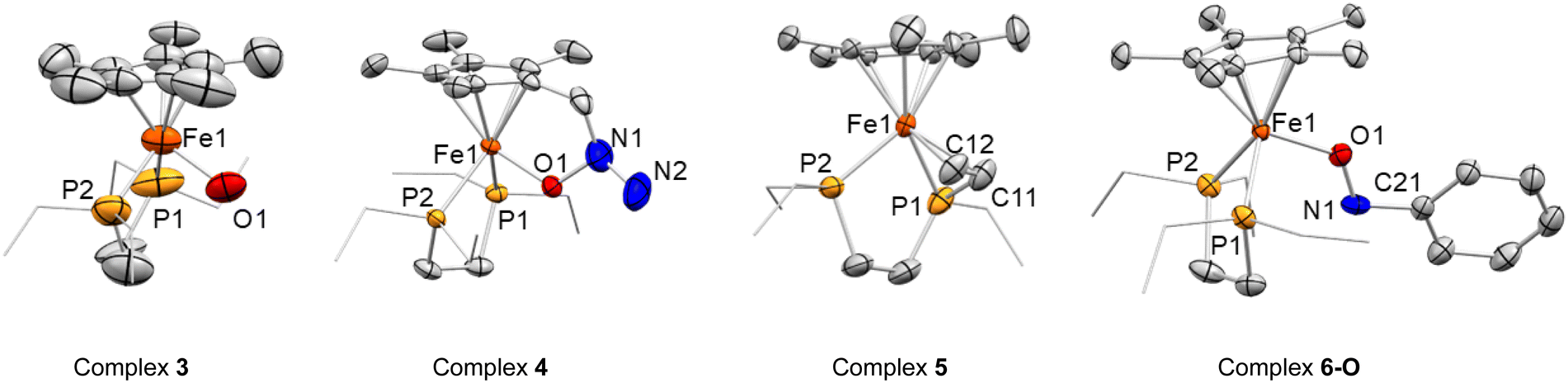 | ||
| Fig. 3 ORTEP of [FeIII(depe)(Cp*)(OH)][BArF] (3), [FeIII(depe)(η5,κ1O–C5Me4CH2N(NH2)O)][BArF] (4), [FeII(κ2P:κ2C-depe′)(Cp*)][BArF] (5) and [FeII(depe)(Cp*)(κ1O–C6H5NO)][BArF] (6-O). Hydrogen atoms and [BArF]− counterions have been excluded, and ethyl groups are represented in wireframe format for clarity. Thermal ellipsoids are set at 50% probability except for complex 3 where thermal ellipsoids are set at 50% probability due to the quality of the crystals. | ||
The first product, [FeIII(depe)(Cp*)(OH)][BArF] (3), is an iron(III)-hydroxido complex, that we attributed to the formation of a very reactive FeIV = O species from N2O cleavage, which would subsequently abstract a hydrogen atom from other molecules.44,45 The second crystalline product, [FeIII(depe)(η5,κ1O–C5Me4CH2N(NH2)O)][BArF] (4), represents an unprecedented cooperative ligand–metal activation product of N2O, yielding an Fe(III) complex in which nitrous oxide has been formally hydrogenated towards a tethered hydrazinyl oxide (−O–N(R)–N(H)2) moiety.
Although one of the incorporated hydrogen atoms seems to originate from a methyl group of the Cp* ligand, the second one probably derives from the other complex, as suggested by the crystallization of [FeII(κ2P:κ2C-depe′)(Cp*)][BArF] (5), where depe′ denotes a diphosphine ligand in which one of the ethyl arms has been dehydrogenated to a vinyl group, that in turn coordinates to Fe(II) in a κ2-fashion. The mechanism of formation of these three exotic species has been investigated by computational means (vide infra), which suggests a complex manifold of reactivities associated with the activation of nitrous oxide.
Attempts to isolate these products in analytically pure form were unsuccessful. Indeed, independent microcrystalline samples isolated from different reaction mixtures were analyzed by X-band EPR spectroscopy at 77 K, always showing the presence of both 3 and 4 (Fig. 2). Simulation of the EPR spectrum of a frozen solution (THF:Et2O = 1:1) of these crystals yielded two distinct components in a 1:1 ratio. The first component (gx = 2.194; gy = 2.053; gz = 2.004) is attributed to compound 3 (Fig. 2), consistent with its formation in the reaction of 2 with nitrobenzene (vide infra). The second component (gx = 2.377, gy = 2.069; gz = 1.994) is associated with compound 4. Nonetheless, we cannot completely rule out the formation of other minor species with S > ½ species, which may not be observed at the temperature used for our measurements (77 K).
The solid-state molecular structure of 3 (Fig. 3) shows a piano-stool cationic iron complex, coordinated by the Cp* ligand, the two phosphorus atoms of the depe and a hydroxido ligand and accompanied by [BArF]− as a counterion. The relatively long Fe–O bond distance (2.030(6) Å) is more consistent with an Fe(III)–OH motif,46–50 rather than an Fe(IV) = O complex, for which a shorter Fe–O length would be expected. Indeed, Density Functional Theory (DFT) calculations at the RI-BP86-D3(BJ)/def2-TZVP level confirm the much longer Fe(III)–OH distance in 3 (1.858 Å) than in the possible Fe(IV) = O species (1.695 Å).The formulation of 3 as an Fe(III)–OH complex is further supported by the X-band EPR spectra from the reaction mixture and from the mixed crystallized material obtained from it. Not surprisingly, our DFT calculations indicate that the unpaired electron in 3 is mainly located at the iron center (computed spin density of 0.93e).
In complex 4, the C–CH2 bond is slightly elongated (ca. 1.506(6) Å) in comparison with the other methyl groups of the Cp* ring, though the distance between the Cp* centroid and the iron center (1.763(4) Å) is comparable to that of the other Fe(III) compound reported herein (3, cf. 1.764(14) Å), though slightly longer than in Fe(II) species 2, 5 and 6 (within the 1.72 to 1.74 Å range). The Fe–O bond distance of 1.844(2) Å falls in the reported range for single bonds between Fe(III) and –OR.46–50
The two hydrogen atoms of the newly formed NH2 group in complex 4 could be unambiguously located in the Fourier electron density map derived from X-ray diffraction studies of different crystal samples. This is also consistent with a N–N distance of 1.497(7) Å, common for a single bond, the presence of a single O–N bond (1.421(5) Å) and the overall charge at the molecule. It is important to highlight that compound 4 represents an unprecedented product of N2O activation and demonstrates the active participation of the Cp* ligand to make it possible.
Complex 5 also exhibits a piano-stool conformation, but besides the two phosphorus atoms, the iron center is coordinated to a new vinyl fragment from the formal dehydrogenation of an ethyl arm. This results in a shorter C–C bond distance (1.404(6) Å), as well as short Fe–CH and Fe–CH2 bond lengths of 2.0566(4) and 2.097(4) Å, respectively. Other angles and distances follow similar patterns and orders compared to those found in 1 and 2. Once more, among the plausible scenarios that would in principle fit with the hydrogen-free model obtained from X-ray diffraction studies for compound 5, DFT calculations supported the aforesaid formulation. In particular, the computed Fe(II)–CH and Fe(II)–CH2 distances of 2.059 Å and 2.101 Å concur quite well with those derived from X-ray diffraction (2.057 and 2.097 Å, respectively).
The unique and complex reactivity discussed with N2O prompted us to investigate the behaviour of 2 with nitrobenzene (C6H5NO2). Addition of the latter to a C6D6 suspension of 2 at 25 °C results in the very slow consumption of the starting material. However, by warming up the reaction mixture to 50 °C, a complete consumption of 2 together with a change of colour to deep blue is observed over the course of 24 h. The 31P{1H} NMR spectrum of the reaction mixture shows the appearance of new resonances around 81.08 and 89.09 ppm (Fig. S25). Once again, two different types of crystals co-crystallize from the vapor diffusion of pentane into a deep blue Et2O solution of the reaction mixture, over the course of 2 days at −35 °C. On the one hand, we obtained orange blocks corresponding to the already described Fe(III)–OH complex, 3. The formation of 3 was also independently confirmed by X-band EPR spectroscopy of the reaction mixture at 77 K (Fig. S77). Besides, very small blue-green plates crystallized as well, which allowed us to determine the overall connectivity (Fig. S69), corresponding to the formation of the Fe(II) nitrosobenzene adduct [FeII(depe)(Cp*)(κ1N–C6H5NO)][BArF], 6-N.
The concomitant formation of these two species suggests a bimolecular activation of the nitrobenzene, which is indeed supported by DFT calculations (vide infra). However, this results in the isolation of 6-N in pure form being challenging, thereby we attempted its independent synthesis by the addition of nitrosobenzene (C6H5NO) to a C6D6 suspension of 2 at 25 °C. Immediately after addition, the suspension acquired the expected deep blue colour, with the presence of oil due to the sparing solubility of the products. Multinuclear NMR spectroscopy confirmed the formation of 6-N in the reaction mixture, together with other species. Single crystals could be grown from the vapor diffusion of pentane into an Et2O solution of the crude. The grown crystals, however, showed the formation of a linkage isomer of 6-N, the O-coordinated nitrosobenzene adduct [Fe(depe)(Cp*)(κ1O–C6H5NO)][BArF], 6-O. The computational studies described below are consistent with the presence of the two isomers, which are almost thermoneutral and may rapidly interconvert (vide infra). Crystals of isomer 6-O (Fig. 3) were of high quality and further confirmed the formation of the nitrosobenzene adduct. The structure is characterized by an Fe–O bond length of 1.833(3) Å, shorter than the one found in 3. As foreseen, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance (1.300(5)Å) is longer than in free nitrosobenzene (1.223 Å)51 due to the interaction of the lone pair of electrons at the oxygen atom with the metal and the backdonation from the iron fragment to the π*(NO).
O bond distance (1.300(5)Å) is longer than in free nitrosobenzene (1.223 Å)51 due to the interaction of the lone pair of electrons at the oxygen atom with the metal and the backdonation from the iron fragment to the π*(NO).
Computational studies
The formation of species 3–5 in the reaction of complex 2 with N2O was explored by means of DFT calculations at the RI-BP86-D3(BJ)/def2-SVP level (Fig. 4, top). First, the process begins with the displacement of the N2 ligand by N2O to form the Fe(II) species [Fe]–ON2. From this cationic intermediate, a H-transfer from a phosphine-ethyl group of the naked [Fe(depe)Cp*]+ complex 2′ would take place, in a slightly exergonic process (ΔG = −1.8 kcal mol−1), to produce complex 5′ along with the Fe(III) radical-cation [Fe]–ON2H (the analogous reaction involving a solvent molecule, i.e. Et2O, as a H-donor is highly endergonic, ΔE = +27.0 kcal mol−1, and therefore, unlikely). From this species, an intramolecular H-migration from a methyl group of the Cp* ligand occurs through transition state TS1 with a relatively low barrier of 19.4 kcal mol−1, consistent with a process occurring at room temperature.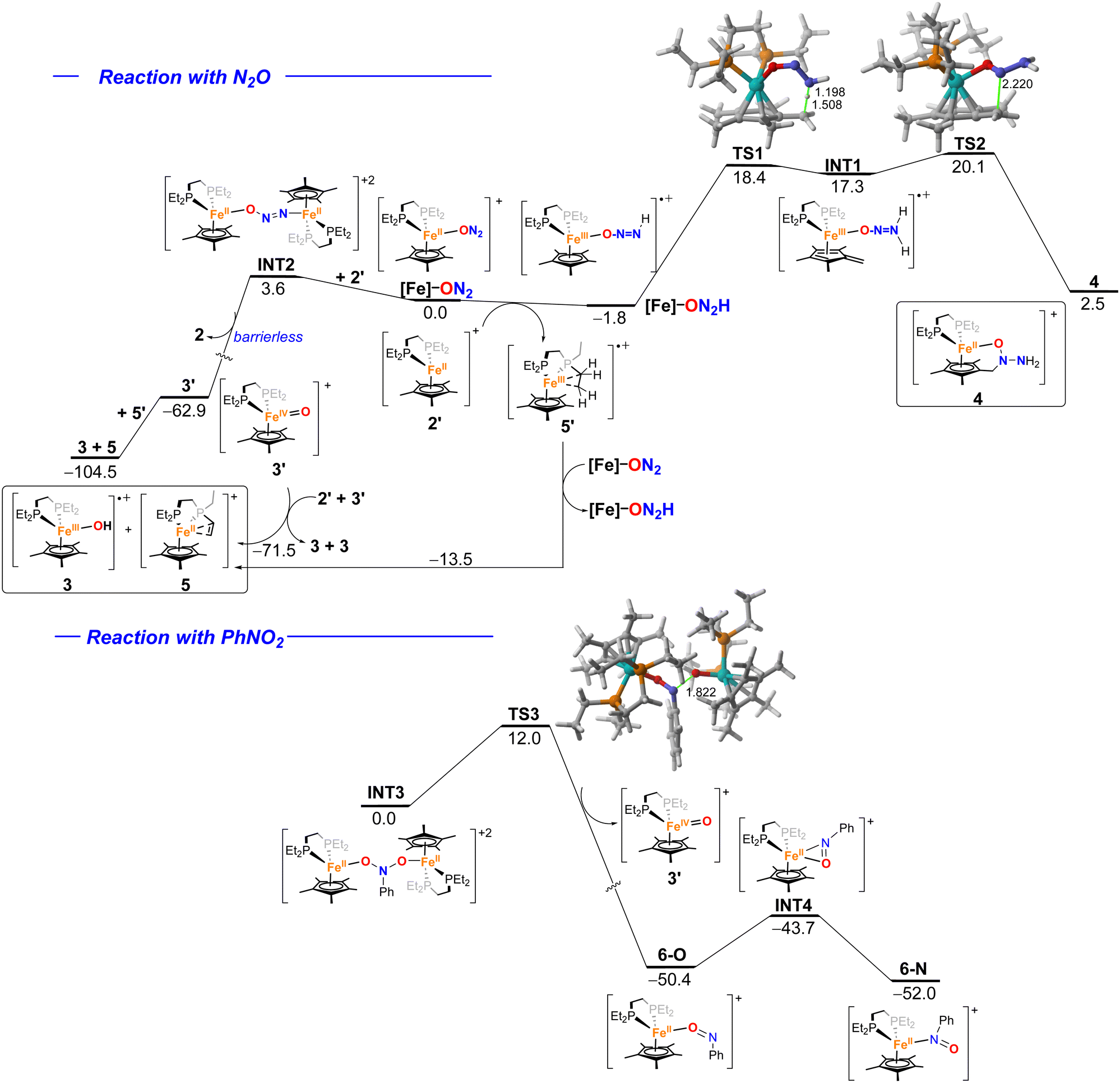 | ||
| Fig. 4 Computed reaction profiles for the reaction of 2 with N2O (top) and nitrobenzene (bottom). Relative free energies (ΔG, at 298 K) and bond distances are given in kcal mol−1 and angstroms, respectively. All data have been computed at the RI-BP86-D3(BJ)/def2-SVP level. | ||
This step leads to the formation of the radical-cation INT1 whose unpaired electron is mainly located at the transition metal and also at the CH2 fragment (computed spin density at carbon of 0.36e). This directs the subsequent formation of the observed C–N bond, which takes place viaTS2 with a rather low barrier of only 2.8 kcal mol−1 leading to the observed complex 4 in a strongly exergonic reaction which compensates for the endergonicity computed for the previous step driving the transformation forward.
The formation of the hydroxido species 3 would also occur from the initially formed [Fe]–ON2 complex. In this case, instead of a H-transfer, the reaction of this species with 2′ leads to the slightly endergonic formation of bimetallic species INT2, where the ON2 ligand binds two different [Fe(depe)Cp*]+ transition metal fragments.16,52 From this intermediate, the oxo-complex Fe(IV) = O 3′ is formed with concomitant release of a molecule of 2 in an essentially barrierless process (see Fig. S74 in the SI).
From 3′, an intermolecular H-transfer from 5′ would lead to the formation of both the hydroxido complex 3 and species 5, in a process highly favored thermodynamically (ΔG = −41.6 kcal mol−1). Alternatively, complex 5 could also derive directly from 2′ upon the abstraction of two H-atoms by two molecules of 3′ (therefore leading to two molecules of 3 as well). This alternative process is also highly exergonic (ΔG = −71.5 kcal mol−1) and seems to be favored over the 3′ + 5′ → 3 + 5 reaction. Moreover, 5 might also result from 5′ upon H-abstraction from a new molecule of the [Fe]–ON2 complex. Although exergonic (ΔG = −13.5 kcal mol−1), this process seems non-competitive as compared to the above-mentioned reactions.
Similar to the reaction with N2O, the process involving nitrobenzene also begins with the formation of the analogous bimetallic complex INT3, where the PhNO2 acts as a bridging ligand between two different [Fe(depe)Cp*]+ fragments (Fig. 4, bottom). In this case, the N–O bond rupture is not barrierless but proceeds through the transition state TS3, with a low barrier of only 12.0 kcal mol−1, in a highly exergonic (ΔG = −50.4 kcal mol−1) transformation leading to the Fe(IV)-oxo complex 3′ and the cationic nitrosobenzene complex 6-O. Our calculations indicate that the experimentally observed isomer 6-N, where the nitroso ligand binds the transition metal through its nitrogen atom, is ca. 2 kcal mol−1 more stable than 6-O. This ligand slippage process takes place via intermediate INT4, lying only 6.7 kcal mol−1 above 6-O, where both the nitrogen and oxygen atoms bind the transition metal. This confirms that the 6-O → 6-N conversion is feasible under the experimental conditions used and therefore consistent with the crystallization of both isomers during our experimental studies.
Catalytic investigations
After demonstrating the capacity of compound 2 to activate N2O and nitrobenzene in several cooperative ways, we decided to explore its implementation into catalytic regimes. In fact, the proposed formation of the iron(IV)-oxo complex (3′) upon reactivity of 2 with N2O (see Fig. 4) offers an ideal platform for studying catalytic oxygen-atom-transfer (OAT) reactions.25,53 We first explored the deoxygenation of N2O by reaction with 20 equiv. of pinacolborane (HBpin) in different solvents at 25 °C (Table 1). Gratifyingly, in THF and Et2O solutions, the conversion is complete over the course of 6 h (entries 1 and 2). No formation of Bpin(OH) is observed, with all reagents being transformed into the corresponding bis-boryloxide O(Bpin)2. In toluene suspension, the reaction is not complete even after 24 h, probably due to the low solubility of the pre-catalyst and reaction intermediates (entry 4). No conversion is observed in dichloromethane solution (entry 3), as a result of the instability of 2 in this solvent, which was confirmed separately. Similarly, no conversion was observed in the absence of catalyst or using the organometallic fragments [FeCl2(depe)2] or [FeCp*2] (entries 8–10).|
|
||||
|---|---|---|---|---|
| Entry | [cat] (mol%) | Solvent | T (°C) | Yield (%)a |
| a Conversion to O(Bpin)2 was calculated via11B{1H} NMR spectroscopy using BPh3 as an internal standard. b The conversion was already complete after 6 h of reaction. | ||||
| 1 | 2 (5) | Et2O | 25 | 100b |
| 2 | 2 (5) | THF | 25 | 100b |
| 3 | 2 (5) | CH2Cl2 | 25 | 0 |
| 4 | 2 (5) | Toluene | 25 | 89 |
| 5 | 2 (0.5) | Et2O | 25 | 49 |
| 6 | 2 (0.5) | Et2O | 50 | 85 |
| 7 | 2 (0.5) | Et2O | 80 | 67 |
| 8 | — | Et2O | 80 | 0 |
| 9 | [FeCl2(depe)2] (5) | Et2O | 80 | 0 |
| 10 | [FeCp*2] (5) | Et2O | 80 | 0 |
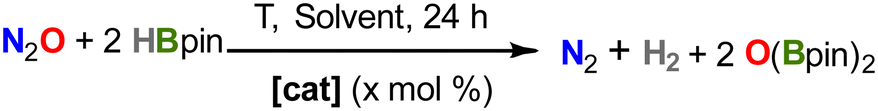
We then decreased catalyst loading to 0.5 mol%, reaching moderate to good yields at variable temperatures, with a maximum of 85% with mild heating at 50 °C (Table 1, entry 6; TON = 171). However, a further increase in the temperature results in lower yields (entry 7), which we attribute to both the lower solubility of the gases and the possibility of catalyst decomposition. The same effect was observed for temperature screening in THF and toluene solvents (see Table S1). Alternatively, using B2pin2 as the reducing agent did not yield any conversion to O(Bpin)2 under our optimized conditions and using 2 as a precatalyst.
Interestingly, we noticed a distinctive mechanistic feature that contrasts with most prior studies. In particular, it seems that this system escapes from the common hydridic mechanism involving oxygen atom insertion into a metal–hydride2,8–13,22,54–56 (or M–C, as seen in other OAT reactions).31 In line with this, HBpin does not react with 2 to produce any potentially reactive Fe–hydride. Indeed, 2 is inert towards HBpin in all solvents used even under harsher conditions, as confirmed by multinuclear NMR experiments (Fig. S30 and S31), while it directly transforms in the presence of N2O (vide supra). However, we do observe the formation of distinctive low-frequency hydridic signals during catalysis. More precisely, 31P{1H} NMR monitoring shows the formation of a diamagnetic iron complex 7 during catalysis, with a chemical shift at δ = 91.1 ppm (Fig. S34 and S35) that correlates to a low-frequency 1H triplet at −9.21 ppm (2JHP = 67.4 Hz). This suggests the formation of the iron-bishydride [FeIV(depe)(Cp*)(H)2][BArF] (7) during the course of the catalytic experiments (Fig. S36). We could corroborate its structure by X-ray diffraction analysis (Fig. S70), which compares well with analogous species reported by the groups of Valerga and LaPinte with different diphosphine ligands.36,37
To test the catalytic competence of dihydride 7, we attempted its independent synthesis, which was achieved by the reaction of 2 with 1 bar of H2 (Fig. S16–S24, Scheme 3). In fact, this experiment suggests that this bis-hydride may form during catalysis due to the production of H2 from the deoxygenative reaction of N2O with HBpin (Table 1). However, this independent synthesis does not lead to pure 7. EPR studies of the reaction mixture at 77 K also showed the concomitant formation of a paramagnetic species with S = ½, suggesting also the potential formation of an Fe(III)-hydride (Fig. S76). The obtained EPR spectrum compares well with previously reported Fe(III)-hydrides by Peters and co-workers using the dppe ligand,36 supporting our postulated formation of [FeIII(depe)(Cp*)H][BArF] (B).
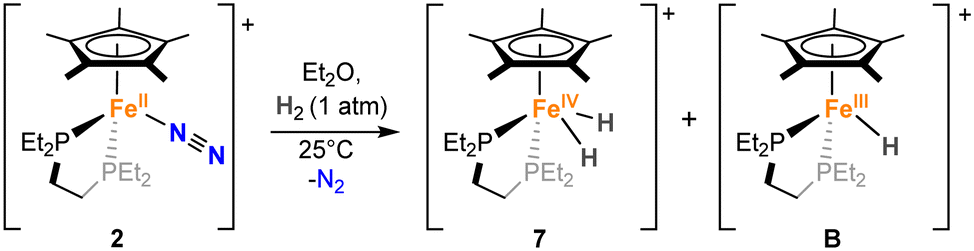 | ||
| Scheme 3 Reaction of 2 with H2 to access iron–hydride complexes [FeIV(depe)(Cp*)(H)2][BArF] (7) and [FeIII(depe)(Cp*)(H)][BArF] (B) (the BArF anion has been omitted in the scheme for clarity). | ||
In any case, the mixture of hydride complexes 7 and B is inactive towards N2O reduction in the absence of HBpin and we therefore attribute their formation to off-cycle species during catalysis. Although at this stage we cannot yet provide a full mechanistic catalytic picture, we postulate that formation of hydrides 7 and B is reversible, giving access to 2, which readily reacts with N2O to form the oxo complex 3′. Then, instead of intramolecularly abstracting a hydrogen atom, the oxo complex reacts with HBpin, possibly leading to hydride B and −OBpin, as proposed by Kaltsoyannis and Mehta.28 The −OBpin anion may deprotonate B to regenerate 2 and form HOBpin, which readily reacts with HBpin to release H2 (explaining the appearance of 7), yielding O(Bpin)2.
We also investigated the catalytic reduction of nitrobenzene by HBpin, which has been reported only recently for two Fe complexes.25,57 In a prototype experiment, a J-Young NMR tube was charged with precatalyst 2, HBpin (5 equiv.) and solvent (0.5 mL), and the reactants were mixed thoroughly. To this mixture, nitrobenzene was added and the reaction was monitored by multinuclear NMR spectroscopy. The reaction proceeds smoothly in Et2O even at room temperature to produce the corresponding N-borylated amine, reaching completion in 30 hours when using 5 mol% catalyst loading (Table 2, entry 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [cat] (mol%) | Solvent | T (°C) | t | Yield (%)a |
| a Yield calculated via11B{1H} NMR spectroscopy using BPh3 as an internal standard. b 94% isolated yield after derivatization to anilinium chloride. | |||||
| 1 | 2 (5) | C6D6 | 25 | 30 h | 65 |
| 2 | 2 (5) | Et2O | 25 | 30 h | 100 |
| 3 | 2 (5) | C6D6 | 50 | 5 h | 100 |
| 4 | 2 (5) | C6D6 | 80 | 30 min | 100 |
| 5 | 2 (0.1) | C6D6 | 80 | 30 min | 81 |
| 6 | 2 (0.1) | C6D6 | 80 | 4 h | 100b |
| 7 | — | C6D6 | 80 | 30 h | 0 |
| 8 | [FeCl2(depe)2] (5) | C6D6 | 80 | 16 h | 10 |
| 9 | [FeCp*2] (5) | C6D6 | 80 | 16 h | 0 |
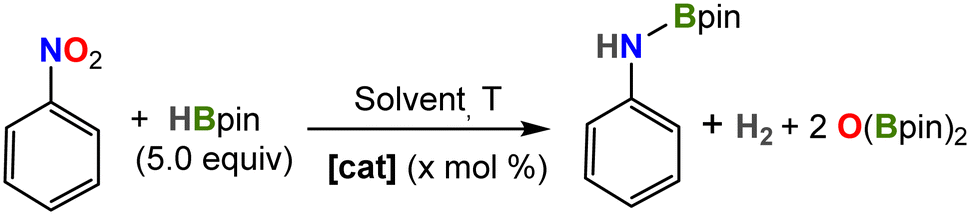
The reaction proceeded at a slower rate in benzene (4 days to completion at 25 °C likely due to reduced solubility); however, the reaction was complete after only 30 min upon heating at 80 °C (entry 4). Indeed, this allowed us to decrease catalyst concentration to 0.1 mol%, reaching full conversion in only 4 hours (entry 6), thereby surpassing the efficiency of the two prior iron systems in terms of turnover number (TON = 1000) by an order of magnitude.25,57 In contrast, there was no conversion in the absence of catalyst or using [FeCp*2], while minimal conversion was observed for precursor [FeCl2(depe)2].
It is worth noting that over the course of the reaction, the mixture became green-blue, as observed in the reaction of 2 with nitrobenzene, suggesting the formation of the nitrosobenzene adduct 6-N as an intermediate of the catalytic transformation. Furthermore, even though the 1H NMR spectra of the reaction mixtures show the formation of H2 (δ = 4.56 ppm), the presence of the aforesaid Fe(IV)-bis hydride 7 was not observed, suggesting that the reaction with nitrobenzene is faster (Fig. S58–S70). Moreover, the isolation of 3 from the reaction of 2 with both nitrobenzene and N2O separately suggests that both catalytic transformations involve common intermediates and therefore proceed through related mechanisms, as will be disclosed in due course.
Conclusions
We have shown that the Cp* ligand in complex [FeII(depe)(Cp*)N2][BArF] (2) stabilizes the metal centre in three different oxidation states (II, III, IV) while playing an active role in the activation of N2O through an unprecedented mechanism. This metal–ligand cooperation pathway results in an exotic Cp*-tethered hydrazinyl oxide (−O–N(R)–N(H)2) moiety which has been structurally characterized and whose formation has been rationalized by DFT calculations. Nonetheless, an alternative bimetallic mechanism involving two iron fragments competes with the latter route. This results in the isolation of the hydroxido complex [FeIII(depe)(Cp*)(OH)][BArF] (3), which points towards the formation of a very reactive Fe(IV) oxo complex (3′) that readily abstracts a hydrogen atom from one ethyl group of the diphosphine ligand, eventually leading to the dehydrogenated complex 5.The bimetallic activation of the N2O molecule is directly related to the N–O cleavage in nitrobenzene (C6H5NO2). Herein, bimetallic cooperation yields two complexes, the corresponding nitrosobenzene adduct (6) and, once more, the highly reactive Fe(IV) intermediate (3′) that rapidly converts into the hydroxido complex 3via H-atom abstraction. Despite the reactive nature of 3′, we have demonstrated that 2 can be used as a precatalyst for O-transfer reactions from N2O and nitrobenzene to HBpin, nicely adding to the still reduced number of catalytic systems based on first-row transition metals competent for these transformations. Overall, the results presented in this work show how an apparently simple ligand framework may offer a rather complex manifold of reactivity that results in an unprecedented mechanism for nitrous oxide activation and for its implementation in catalytic regimes.
Author contributions
N. J. carried out the experimental work: synthesis and characterization of new complexes, reactivity studies and catalysis. H. C. performed X-ray diffraction studies. N. J and E. S. carried out EPR experiments. I. F. carried out computational investigations. J. C. supervised the overall work. N. J. and J. C. wrote the manuscript with participation of all authors.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2475085–2475092 contain the supplementary crystallographic data for this paper.58a–hData relating to the synthesis and characterization of new compounds, general methods, NMR and EPR spectra, crystal structure determination and computational studies are available in the supplementary information (SI). The data supporting this article have been included as part of the SI. The corresponding characterization, reactivity and catalysis raw data files are available in the Digital.CSIC open repository (DOI: https://doi.org/10.20350/digitalCSIC/17882). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07656k.
Acknowledgements
This work was funded by the European Union (ERC Consolidator Grant, BiMetalGAS, 101169746), the Spanish MCIU/AEI/10.13039/501100011033 (Grants PID2022-139318NB-I00, RED2022-134287-T) and Fundación Ramón Areces (Ayudas a la Investigación en Ciencias de la Vida y de la Materia). N. J. acknowledges the Spanish Ministry of Science, Innovation and Universities for a Juan de la Cierva postdoctoral fellowship (JDC2022-049349-I). We also want to thank Dr Marta Rosello Merino for the helpful discussions regarding the NMR characterization of 7.Notes and references
- D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120–123 CrossRef CAS PubMed.
- R. Zeng, M. Feller, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 5720–5723 CrossRef CAS.
- X. Wu, J. Du, Y. Gao, H. Wang, C. Zhang, R. Zhang, H. He, G. Lu and Z. Wu, Chem. Soc. Rev., 2024, 53, 8379–8423 Search PubMed.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- S. Lee, S. J. Park and J. Lee, Valorisation of Greenhouse Gas for Commodity Chemical Production–Nitrous Oxide as Oxidising Agent for Heterogeneous Catalysis, ChemSusChem, 2025, 18(12), e202402728 CrossRef CAS PubMed.
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, J. Am. Chem. Soc., 2019, 119(19), 10916–10976 CAS.
- K. Severin, Trends Chem., 2023, 5, 574–576 Search PubMed.
- P. Jurt, A. S. Abels, J. J. Gamboa-Carballo, I. Fernández, G. Le Corre, M. Aebli, M. G. Baker, F. Eiler, F. Müller, M. Wörle, R. Verel, S. Gauthier, M. Trincado, T. L. Gianetti and H. Grützmacher, Angew. Chem., Int. Ed., 2021, 60, 25372–25380 CrossRef CAS PubMed.
- J. Bösken, R. E. Rodríguez-Lugo, S. Nappen, M. Trincado and H. Grützmacher, Chem. Eur J., 2023, 29(20) DOI:10.1002/chem.202203632.
- M. Kjellberg, A. Ohleier, P. Thuéry, E. Nicolas, L. Anthore-Dalion and T. Cantat, Chem. Sci., 2021, 12, 10266–10272 RSC.
- I. Ortega-Lepe, P. Sánchez, L. L. Santos, P. Lara, N. Rendón, J. López-Serrano, V. Salazar-Pereda, E. Álvarez, M. Paneque and A. Suárez, Inorg. Chem., 2022, 61, 18590–18600 CrossRef CAS.
- R. Zeng, M. Feller, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2018, 140, 7061–7064 CrossRef CAS PubMed.
- T. L. Gianetti, S. P. Annen, G. Santiso-Quinones, M. Reiher, M. Driess and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 128, 1886–1890 CrossRef.
- S. H. Dewick, T. M. Hood, Y. Han, S. Huband and A. B. Chaplin, Catal. Sci. Technol., 2025, 15, 4126–4129 RSC.
- S. T. Nappen, J. J. Gamboa-Carballo, E. Tschanen, F. Ricatto, M. D. Wörle, A. Thomas, M. Trincado and H. Grützmacher, Angew. Chem., Int. Ed., 2025, 64(25) DOI:10.1002/anie.202502616.
- J. G. Andino and K. G. Caulton, J. Am. Chem. Soc., 2011, 133(32), 12576–12583 Search PubMed.
- A. Y. Verat, H. Fan, M. Pink, Y.-S. Chen and K. G. Caulton, Chem. Eur J., 2008, 14, 7680–7686 Search PubMed.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- R. J. M. Gebbink, M.-Etienne. Moret. Non-noble metal catalysis : molecular approaches and reactions, Wiley-VCH, 2019 Search PubMed.
- S. Ni, F. Le Vaillant, A. Mateos-Calbet, R. Martin and J. Cornella, J. Am. Chem. Soc., 2022, 144, 18223–18228 CrossRef CAS PubMed.
- F. Le Vaillant, A. Mateos Calbet, S. González-Pelayo, E. J. Reijerse, S. Ni, J. Busch and J. Cornella, Nature, 2022, 604, 677–683 Search PubMed.
- J. Bermejo, I. Ortega-Lepe, L. L. Santos, N. Rendón, J. López-Serrano, E. Álvarez and A. Suárez, Chem. Commun., 2024, 60, 1575–1578 RSC.
- H. Zhang, J. Rodrigalvarez and R. Martin, J. Am. Chem. Soc., 2023, 145, 17564–17569 CrossRef CAS.
- T. M. Hood, R. S. C. Charman, D. J. Liptrot and A. B. Chaplin, Angew. Chem., Int. Ed., 2024, 63(45) DOI:10.1002/anie.202411692.
- X. Chen, H. Wang, S. Du, M. Driess and Z. Mo, Angew. Chem., Int. Ed., 2022, 61, e202114598, DOI:10.1002/anie.202114598.
- S. Saito, H. Ohtake, N. Umezawa, Y. Kobayashi, N. Kato, M. Hirobe and T. Higuchi, Chem. Commun., 2013, 49, 8979–8981 RSC.
- J. S. Stanley, X. S. Wang and J. Y. Yang, ACS Catal., 2023, 13(19), 12617–12622 CrossRef CAS.
- B. V. IJzendoorn, R. Lister-Roberts, N. Kaltsoyannis and M. Mehta, J. Am. Chem. Soc., 2025, 147, 30317–30325 CrossRef PubMed.
- Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19473–19479 CrossRef CAS PubMed.
- Z. Liu, X. Li, Z. Wang, H. Mu, B. Xiao, W. Xue, J. Zhou and Z. Dong, Angew. Chem., Int. Ed., 2025, e202515638, DOI:10.1002/anie.202515638.
- A. Mateos-Calbet, P. C. Bruzzese, M. A. Mermigki, A. Schnegg, D. A. Pantazis and J. Cornella, J. Am. Chem. Soc., 2025, 147, 19438–19443 CrossRef CAS PubMed.
- H. Corona, M. Pérez-Jiménez, F. de la Cruz-Martínez, I. Fernández and J. Campos, Angew. Chem., Int. Ed., 2022, 61(40) DOI:10.1002/anie.202207581.
- C. S. Durfy, J. A. Zurakowski and M. W. Drover, Angew. Chem., Int. Ed., 2025, 64(9) DOI:10.1002/anie.202421599.
- J. A. Zurakowski and M. W. Drover, Chem. Commun., 2023, 59, 11349–11352 RSC.
- K. Jaiswal, N. Malik, B. Tumanskii, G. Ménard and R. Dobrovetsky, J. Am. Chem. Soc., 2021, 143, 9842–9848 CrossRef CAS PubMed.
- D. J. Schild, M. W. Drover, P. H. Oyala and J. C. Peters, J. Am. Chem. Soc., 2020, 142, 18963–18970 CrossRef CAS PubMed.
- H. K. Sharma, I. Haiduc, K. H. Pannell, Transition Metal Complexes of Germanium, Tin and Lead. in The Chemistry of Organic Germanium, Tin and Lead Compounds, ed. Z. Rappoport, Wiley, Chichester, 2009, pp. 2 Search PubMed.
- M. Jimenez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 1994, 13, 3330–3337 CrossRef CAS.
- J. A. Zurakowski, K. R. Brown and M. W. Drover, Inorg. Chem., 2023, 62, 7053–7060 CrossRef CAS PubMed.
- K. Jonas, P. Klusmann and R. Goddard, Z. Naturforsch. B Chem. Sci., 1995, 50, 394–404 CrossRef.
- A. K. Eckhardt, M. L. Y. Riu, M. Ye, P. Müller, G. Bistoni and C. C. Cummins, Nat. Chem., 2022, 14, 928–934 CrossRef CAS PubMed.
- C. Roger, P. Hamon, L. Toupet, H. Rabaa, J. Y. Saillard, J. R. Hamon and C. Lapinte, Organometallics, 1991, 10, 1045–1054 CrossRef CAS.
- A. De La, J. Leal, M. Jimenez Tenorio, M. Carmen Puerta and P. Valerga, Organometallics, 1995, 14(8), 3839–3847 CrossRef.
- J. England, Y. Guo, E. R. Farquhar, V. G. Young, E. Münck and L. Que, J. Am. Chem. Soc., 2010, 132, 8635–8644 CrossRef CAS PubMed.
- M. Puri and L. Que, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed.
- D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS PubMed.
- W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2007, 129, 15128–15129 CrossRef CAS PubMed.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS.
- C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young, G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 2556–2567 CrossRef CAS.
- C. E. MacBeth, A. P. Golombek, V. G. Young, Y. C. Jr, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938–941 CrossRef CAS PubMed.
- R. Boese, D. Bläser, M. Nussbaumer and T. M. Krygowski, Struct. Chem., 1992, 3, 363–368 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2007, 40(7), 522–531 CrossRef CAS PubMed.
- H. Xie, Y. Zhang, C. Xiang, Y. Li, T. Fan, Q. Lei and W. Fang, Dalton Trans., 2018, 47, 15324–15330 RSC.
- J. A. Luque-Urrutia and A. Poater, Inorg. Chem., 2017, 56, 14383–14387 CrossRef CAS PubMed.
- S. Escayola, M. Solà and A. Poater, Inorg. Chem., 2020, 59, 9374–9383 CrossRef CAS PubMed.
- T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, M. Chui, M. Temprado, J. S. Silvia, A. F. Cozzolino, D. Tofan, A. Velian, C. C. Cummins, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2013, 135(30), 11357–11372 CrossRef CAS.
- E. Pocock, M. Diefenbach, T. M. Hood, M. Nunn, E. Richards, V. Krewald and R. L. Webster, J. Am. Chem. Soc., 2024, 146, 19839–19851 CrossRef CAS PubMed.
- (a) CCDC 2475085: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jgs; (b) CCDC 2475086: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jht; (c) CCDC 2475087: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jjv; (d) CCDC 2475088: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jkw; (e) CCDC 2475089: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jlx; (f) CCDC 2475090: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jmy; (g) CCDC 2475091: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jnz; (h) CCDC 2475085: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p2jp0.
| This journal is © The Royal Society of Chemistry 2026 |