
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of calix[4]arenes with C–C axial and inherent chirality via palladium/chiral norbornene cooperative catalysis
Yiming
You
a,
Hongwei
Cheng
a,
Xiao
Huang
a,
Peng
Wang
c,
Hengjiang
Cong
 a,
Hong-Gang
Cheng
*a and
Qianghui
Zhou
*ab
a,
Hong-Gang
Cheng
*a and
Qianghui
Zhou
*ab
aHubei Research Center of Fundamental Science-Chemistry, Engineering Research Center of Organosilicon Compounds & Materials (Ministry of Education), Hubei Key Lab on Organic and Polymeric OptoElectronic Materials, College of Chemistry and Molecular Sciences, The Institute for Advanced Studies, Taikang Center for Life and Medical Sciences, State Key Laboratory of Metabolism and Regulation in Complex Organisms, Wuhan University, Wuhan, 430072, China. E-mail: hgcheng@whu.edu.cn; qhzhou@whu.edu.cn
bSchool of Artificial Intelligence, Wuhan University, Wuhan, 430072, China
cState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, China
First published on 4th March 2026
Abstract
The catalytic asymmetric synthesis of inherently chiral calix[4]arenes, a distinct class of chiral macrocycles with significant applications in asymmetric catalysis, chiral recognition and functional materials, remains a formidable challenge. Moreover, strategies for the simultaneous introduction of multiple stereogenic elements within these scaffolds in a single transformation are highly desirable yet underdeveloped. Herein, we report a one-step protocol that concurrently installs both C–C axial and inherent chirality in a calix[4]arene system through palladium/chiral norbornene (Pd/NBE*) cooperative catalysis. This approach provides direct access to a diverse array of five- and six-membered benzo-fused calix[4]arenes (30 examples) with consistently high enantioselectivity and diastereoselectivity. Notably, the obtained products can be readily transformed into monophosphine ligands bearing both C–C axial and inherent chirality, which exhibit excellent stereocontrol in a silver-catalysed asymmetric [3 + 2] cyclization reaction. Preliminary photophysical and chiroptical studies reveal that these dual-chiral calix[4]arenes exhibit promising glum factors, demonstrating their potential as versatile chiral scaffolds for advanced organic optoelectronic materials.
Introduction
Inherent chirality, a concept introduced by Böhmer in 1994, refers to chirality arising from the asymmetric arrangement of achiral substituents within calix[n]arene frameworks.1 This concept has since been extended to other chiral cyclic architectures that distinguished with conventional central, axial, planar, or helical chirality, including calix[n]arenes, saddle-shaped rings, rotaxanes and catenanes, as well as others (Fig. 1A).2 Among these, inherently chiral calix[4]arenes constitute a particularly compelling subclass due to their distinctive three-dimensional cavities, which confer significant potential in asymmetric catalysis, chiral recognition, and advanced materials (Fig. 1B).3 Consequently, the development of efficient and stereoselective synthetic routes to these macrocycles has been a central pursuit. Early landmark achievements, such as McKervey's lipase-catalysed transesterification4 and Tsue's Pd-catalysed intramolecular Buchwald–Hartwig amination,5 established initial approaches but faced limitations in efficiency or enantiocontrol. A significant advance was achieved by Tong, Wang, and co-workers, who developed a series of effective strategies for the assembly of heteracalix[4]aromatics with high enantioselectivity, including asymmetric intramolecular Buchwald–Hartwig amination, SNAr reaction, Suzuki–Miyaura coupling and dynamic kinetic resolution (DKR).6 More recently, catalytic desymmetrization of prochiral calix[4]arene precursors has emerged as a powerful and efficient route. In 2022, Cai and co-workers developed a Pd-catalysed intramolecular C–H arylation using a chiral phosphine–carboxylate ligand, providing fluorenone-derived calix[4]arenes with a notable luminescence dissymmetry factor (glum) (Fig. 1C, i-a).7 Concurrently, Tong, Wang, and co-workers achieved a Pd/chiral phosphoramidite-catalysed sequential 1,5-palladium migration/intramolecular C–H arylation to construct 9H-fluorene-embedded inherently chiral calix[4]arenes (Fig. 1C, i-b).8 Recently, Cera, Pirovano et al. demonstrated an enantioselective gold(I)-catalysed intramolecular hydroarylation of alkynes, affording phenanthrene-containing calix[4]arenes in high yields and enantioselectivities (Fig. 1C, i-c).9 Parallel advances in directed ortho C–H functionalization have further enriched the synthetic toolbox. For example, Chen and co-workers developed an elegant amine-directed electrophilic sulfenylation strategy using a chiral sulfide catalyst and hexafluoroisopropanol (Fig. 1C, ii-a).10 Subsequently, the Yang and Nan groups realized a chiral phosphoric acid (CPA)-catalysed electrophilic aromatic amination or bromination of phenol/amine-decorated calix[4]arenes (Fig. 1C, ii-b, c),11 while Li, Wang and co-workers accomplished a Rh-catalysed ortho C–H olefination of calix[4]arene-based carboxamides (Fig. 1C, ii-d).12 Complementary organocatalytic desymmetrization approaches have also been successfully applied, including NHC-catalysed esterification by Veselý, Dočekal (Fig. 1C, iii)13 and Yan's CPA-catalysed enantioselective halo-cyclization (Fig. 1C, iv).14 Most recently, annulation strategies have proven highly effective for the construction of inherently chiral calix[4]arenes. In this context, Liu, Yang and Zhou independently disclosed a CPA-catalysed Povarov/aromatization sequence (Fig. 1C, v-a).15 The groups of Niu and Shi concurrently developed a cobalt(III)/Salox-catalysed C–H activation/[4 + 2] annulation with alkynes to simultaneously install both inherent and C–N axial chirality (Fig. 1C, v-c).16 Li, Wang and co-workers realized a Rh-catalysed ortho C–H alkynylation followed by intramolecular [4 + 2] annulation (Fig. 1C, v-b), as well as a Rh-catalysed oxidative [4 + 1] annulation with 1,3-enynes to access calix[4]arenes bearing both inherent and central chirality with excellent stereocontrol (Fig. 1C, vi).12 Despite these impressive advancements, current methodologies remain predominantly focused on the assembly of calix[4]arenes with solely inherent chirality. The concurrent integration of an additional stereogenic element, such as C–C axial chirality, within the same scaffold is largely underdeveloped. Such “dual-chiral” architectures could unlock synergistic functions and novel material properties but pose a formidable synthetic challenge, requiring precise control over multiple stereogenic elements in a single transformation. | ||
| Fig. 1 Catalytic asymmetric approaches to access inherently chiral calix[4]arenes. | ||
Our group has recently demonstrated the power of palladium/chiral norbornene (Pd/NBE*) cooperative catalysis17 (also known as the asymmetric Catellani reaction18) to construct atropisomeric o-terphenyls with 1,2-diaxes19 or ferrocenes with both axial and planar chirality,20 guided by axial-to-axial or axial-to-planar diastereoinduction, respectively. Inspired by these studies, we envisioned that this catalytic platform could be extended to achieve an unprecedented axial-to-inherent diastereoinduction, enabling the one-step assembly of calix[4]arenes bearing both C–C axial and inherent chirality. As depicted in Fig. 1D, we proposed that in the presence of Pd/NBE* cooperative catalysis, the reaction between readily accessible ortho-calix[4]arene-tethered aryl iodide 1 and a bulky β-substituted naphthyl bromide 2 would first generate an axially chiral PdII complex I. This intermediate would then undergo an intramolecular, enantioselective C(sp2)–H activation, where the pre-established axial chirality dictates the formation of the inherent chirality during cyclization, affording the desired dual-chiral product 3. Given the ready accessibility of the starting materials (1 and 2) and the unique axial-to-inherent diastereoinduction mode, this strategy provides a highly efficient platform to access calix[4]arenes with both C–C axial and inherent chirality. However, multiple challenges are also associated with this cascade process: (1) competing side reactions such as direct cyclization of 1 or protodepalladation of intermediate I must be suppressed and (2) the efficiency of the unprecedented axial-to-inherent diastereoinduction is difficult to predict, as it depends critically on controlling the conformational dynamics of the calix[4]arene during the stereodefining C–H activation step. Herein, we report the successful realization of this strategy, providing a direct, one-step synthetic route to a diverse range of benzo-fused calix[4]arenes bearing concurrent C–C axial and inherent chirality with high efficiency and excellent stereocontrol.
Results and discussion
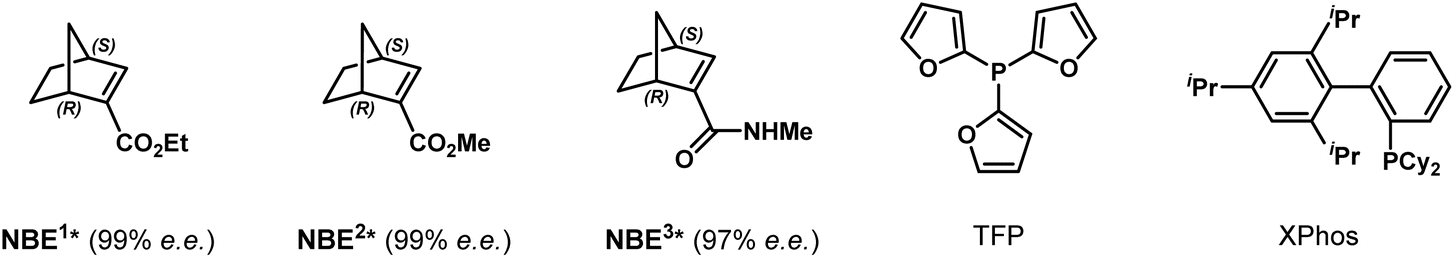
To explore this intriguing process, we commenced our study with a model reaction between ortho-calix[4]arene-tethered aryl iodide 1a and methyl 1-bromo-2-naphthoate 2a (Table 1). Delightfully, under initial conditions using Pd(acac)2 (10 mol%), TFP (tri(furan-2-yl)phosphine) (10 mol%), (1R,4S)-2-ethyl ester-substituted norbornene NBE1* (99% e.e., 30 mol%), pivalic acid (30 mol%), and Cs2CO3 (2.0 equiv.) in toluene (0.1 M) at 80 °C, the desired product 3a with both C–C axial and inherent chiralities was obtained in 60% yield with excellent enantioselectivity (>99%/99% e.e.) and moderate diastereoselectivity (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.) (entry 1). Switching the palladium precursor from Pd(acac)2 to Pd(OPiv)2 enhanced the yield to 71% (entry 2), possibly because the pivalate anion facilitates C–H activation and suppresses competing pathways. Subsequent ligand and NBE* screening identified TFP and NBE1* as the optimal combination (entries 2–6). Diluting the reaction concentration to 0.05 M further improved the yield to 78% while retaining high stereoselectivity (entry 7). Finally, replacing the naphthoate substrate 2a with the more sterically hindered diphenylphosphonyl-substituted analogue 2b afforded product 3b in 81% isolated yield with excellent enantioselectivity (>99% e.e.) and diastereoselectivity (d.r. >19:1) (entry 8). Therefore, the reaction conditions listed in entry 8 were established as the optimal ones for this transformation (see Tables S1–S5 for optimization details).
:1 d.r.) (entry 1). Switching the palladium precursor from Pd(acac)2 to Pd(OPiv)2 enhanced the yield to 71% (entry 2), possibly because the pivalate anion facilitates C–H activation and suppresses competing pathways. Subsequent ligand and NBE* screening identified TFP and NBE1* as the optimal combination (entries 2–6). Diluting the reaction concentration to 0.05 M further improved the yield to 78% while retaining high stereoselectivity (entry 7). Finally, replacing the naphthoate substrate 2a with the more sterically hindered diphenylphosphonyl-substituted analogue 2b afforded product 3b in 81% isolated yield with excellent enantioselectivity (>99% e.e.) and diastereoselectivity (d.r. >19:1) (entry 8). Therefore, the reaction conditions listed in entry 8 were established as the optimal ones for this transformation (see Tables S1–S5 for optimization details).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2 | Pd cat. | L | NBE* | Yieldb (%) | d.r.c | e.e.d (%) |
| a All reactions were performed on a 0.05 mmol scale. b The yield was determined by 19F NMR analysis with 4,4′-difluoro-1,1′-biphenyl as an internal standard. c d.r. was determined by crude 19F NMR analysis. d e.e. was determined by chiral HPLC analysis. e Toluene (0.05 M) was applied. f 2b (1.6 equiv.) was applied. g Isolated yield in parentheses. TFP: tri(furan-2-yl)phosphine. | |||||||
| 1 | 2a | Pd(acac)2 | TFP | NBE1* | 60 | 5:1 |
>99/99 |
| 2 | 2a | Pd(OPiv)2 | TFP | NBE1* | 71 | 6:1 |
>99/99 |
| 3 | 2a | Pd(OPiv)2 | XPhos | NBE1* | 7 | — | — |
| 4 | 2a | Pd(OPiv)2 | PPh3 | NBE1* | 65 | 6:1 |
>99/99 |
| 5 | 2a | Pd(OPiv)2 | TFP | NBE2* | 61 | 6:1 |
>99/99 |
| 6 | 2a | Pd(OPiv)2 | TFP | NBE3* | 5 | — | — |
| 7e | 2a | Pd(OPiv)2 | TFP | NBE1* | 78 | 6:1 |
>99/99 |
| 8e,f | 2b | Pd(OPiv)2 | TFP | NBE1* | 79 (81)g | >19:1 |
>99 |
|
|||||||
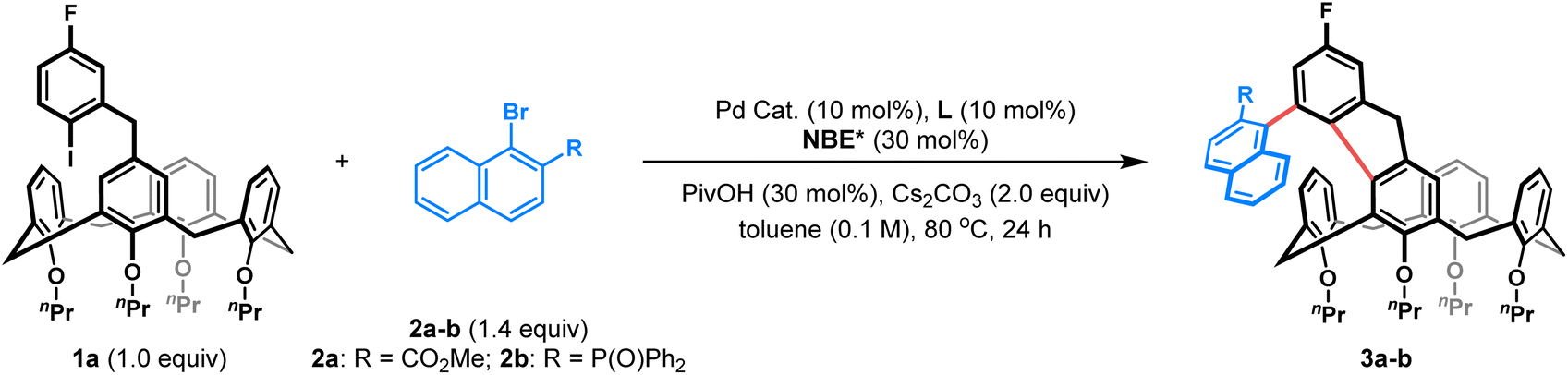
With the optimal reaction conditions established, we first investigated the scope of ortho-calix[4]arene-tethered aryl iodide 1, employing naphthyl bromide 2b or 2c as the fixed reaction partner. As shown in Table 2, varying alkoxyl groups at the lower-rim of calix[4]arenes had a minimal impact on stereoselectivity. Substrates 1b–e, bearing nPr, nBu, Bn or MOM groups, all reacted smoothly to afford the corresponding products 3c–f in 52–81% yields with >99% e.e. and >19:1 d.r. In addition, halogen functionality was also tolerated, as illustrated by the para-bromo-substituted derivative (1f), which afforded product 3g in 74% yield with excellent stereocontrol. Notably, the electronic nature of the aryl iodide tether proved highly adaptable. Aryl iodides with an electron-donating group (e.g., Me, OMe, and NMe2) or electron-withdrawing group (e.g., F, Cl, and CO2Me) all proved to be competent substrates, giving the desired products 3b and 3h–m in 55–81% yields with >99% e.e. and >19:1 d.r. Moreover, a carbazole-fused calix[4]arene 3n was furnished in 48% yield with 95% e.e. and >19:1 d.r. using ortho-amino substituted aryl iodide 1m as the substrate. Furthermore, aryl iodides with a longer tether were also suitable for this approach, providing access to six-membered benzo-fused calix[4]arenes 3o–r in 54–71% yields with >99% e.e. and 10:1 to >19:1 d.r.
| a Reaction conditions: 1 (0.1 mmol, 1.0 equiv.), 2 (0.16 mmol, 1.6 equiv.), Pd(OPiv)2 (10 mol%), TFP (10 mol%), NBE1* (30 mol%), PivOH (30 mol%), Cs2CO3 (2.0 equiv.), and toluene (0.05 M) at 80 °C for 24 h. b 1.5 mmol scale. c 48 h instead of 24 h. d 110 °C instead of 80 °C. e 72 h instead of 24 h. f Yield based on the recovered starting material. |
|---|

|
We next surveyed the scope of the bulky naphthyl bromide 2, with 1b as the fixed coupling partner. A series of 2-diarylphosphinyl-1-naphthyl bromides with either electron-donating substituents (e.g., p-Me, p-OMe, and 3,5-di-tBu) or electron-withdrawing substituents (e.g., p-Cl) on the aryl rings reacted efficiently to yield products 3s–v in moderate to good yields (63–85%). Additionally, 2-naphthyl, 2-thienyl and methyl phosphine oxide-substituted naphthyl bromides also served as suitable arylating reagents to provide the corresponding products 3w–y in 35–81% yields. It is noteworthy that 6-bromo-diphenylphosphine oxide (3z) displayed good reactivity as well. Apart from the phosphonyl groups, other functional handles such as esters (3a and 3A), amides (3B–C), and a sulfonyl group (3D) were all successfully incorporated, affording the desired products in 43–95% yields. Notably, all products in this series were obtained with excellent enantioselectivities (99% e.e.) and good to excellent diastereoselectivities (5.5:1 to >19:1 d.r.). Importantly, the scalability of this approach was demonstrated by a 1.5 mmol-scale experiment, which provided 1.1 g of 3c in 76% yield with >99% e.e. and >19:1 d.r., alongside 88% recovery of the NBE1* cocatalyst.
As the obtained calix[4]arenes with both C–C axial and inherent chiralities are versatile intermediates, their synthetic utility is illustrated by follow-up transformations (Fig. 2A). For instance, reduction of 3c readily afforded the novel phosphine 4, which was preliminarily validated as an effective chiral ligand in a silver-catalysed asymmetric [3 + 2] cyclization reaction21 (Fig. 2B). In addition, nucleophilic substitution of 3c with benzyl bromide led to the formation of fluorene-fused calix[4]arene 5 as a single diastereomer in 53% yield. Oxidation of 3c under air in the presence of KOH produced fluorenone-fused calix[4]arene 6 in 92% yield. The absolute configuration of 6 was unambiously determined via X-ray crystallographic analysis (CCDC 2403829). Subsequently, nucleophilic addition of 6 with phenylmagnesium bromide proceeded smoothly to afford fluorenol-fused calix[4]arene 7 as a single diastereomer in 84% yield. Notably, compounds 5 and 7 each integrate three distinct stereogenic elements: axial, inherent and central chirality, with the configuration of newly generated stereocenter being fully controlled by the pre-existing inherent chirality. Finally, lithium–halogen exchange of 3-bromo-5,5′-di-tert-butyl-2,2′-bithiophene, followed by the addition to fluorenone 6 and subsequent intramolecular Friedel–Crafts cyclization, provided the dithiophene-fused spirofluorene 8 in 51% yield. This compound exhibits promising circularly polarized luminescence (CPL) properties (vide infra).
 | ||
| Fig. 2 (A) Follow-up transformations. Reaction conditions: (a) HSiCl3, Et3N, toluene, 120 °C; (b) nBuLi, BnBr, THF, 0 °C to r.t.; (c) KOH, air, DMSO, 40 °C; (d) PhMgBr, THF, 0 °C to r.t.; (e) 3-bromo-5,5′-di-tert-butyl-2,2′-bithiophene, nBuLi, THF, −78 °C, then AcOH, H2SO4, nhexane/DCM, 60 °C. (B) Synthetic application. | ||
With these novel calix[4]arenes bearing both C–C axial and inherent chirality in hand, we initially examined the photophysical properties of compounds 3c, 3y, 3p and 8 using ultraviolet-visible (UV-vis) absorption and photoluminescence spectroscopy. As shown in Fig. 3a, 3c, 3y, 3p and 8 showed similar profiles, with strong absorption maxima around 275 nm and weaker features near 320 nm. These compounds displayed clearly broadened fluorescence emission bands, with maxima at 431 nm for 3c, 420 nm for 3y, 417 nm for 3p and 434 nm for 8 (Fig. 3b). Moreover, the quantum yields were determined to be 0.241, 0.359, 0.134, and 0.146 for 3c, 3y, 3p, and 8, respectively (Fig. 3c). We next explored the chiroptical properties of these compounds via circular dichroism (CD) and circularly polarized luminescence (CPL) spectroscopy, respectively. As clearly demonstrated by the CD spectra (Fig. 3d), all these compounds exhibited pronounced Cotton effects ranging from 380 to 550 nm (Fig. 3e). Remarkably, CPL experiments indicated that these compounds were all CPL-active, with compound 3p displaying the highest luminescence dissymmetry factor (glum) (3.2 × 10−3 at 411 nm, Fig. 3f), which is comparable to conventional organic small molecules that typically have glum values ranging from 10−5 to 10−3. These results demonstrated the significant potential of dual-chiral calix[4]arene architectures as promising candidates for new chiral organic optoelectronic materials.
 | ||
| Fig. 3 Investigation of photophysical and chiroptical properties. (a) Absorption spectra of 3c, 3y, 3p, and 8 in DCM (5.0 × 10−5 M). (b) Emission spectra of 3c, 3y, 3p, and 8 in DCM (5.0 × 10−5 M). (c) Quantum yields of 3c, 3y, 3p, and 8 in DCM (5.0 × 10−5 M). (d) CD spectra of 3c, 3y, 3p, and 8 in DCM (5.0 × 10−5 M) at r.t. (e) CPL spectra of 3c, 3y, 3p, and 8 in DCM (5.0 × 10−5 M) at r.t. (f) glum value–wavelength curves of 3c, 3y, 3p, and 8. | ||
Conclusions
In summary, we have developed an efficient strategy to access calix[4]arenes with both C–C axial and inherent chirality via Pd/NBE* cooperative catalysis. In this intriguing cascade protocol, the initially introduced axial chirality is orchestrated by Pd/NBE* cooperative catalysis, whereas the subsequent inherent chirality is controlled by the preinstalled axial chirality through an unprecedented axial-to-inherent diastereoinduction process. Using this approach, a diverse range of five- and six-membered benzo-fused calix[4]arenes bearing both C–C axial and inherent chirality were synthesized in a single step with excellent enantioselectivity and diastereoselectivity. These dual-chiral products were readily transformed into new monophosphine ligands, which demonstrated outstanding stereocontrol in a silver-catalysed asymmetric [3 + 2] cyclization reaction. Furthermore, photophysical and chiroptical characterization disclosed promising glum values for these compounds, highlighting their significant potential in developing new chiral organic optoelectronic materials.Author contributions
H.-G. Cheng and Q. Zhou conceived the idea and directed the project. Y. You, H. Cheng, X. Huang and P. Wang performed the experiments under the supervision of H.-G. Cheng and Q. Zhou. H. Cong performed the X-ray crystallographic analysis. Y. You, H.-G. Cheng and Q. Zhou co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The supporting data have been provided as part of the supplementary information (SI). Supplementary information: optimization studies, experimental procedures, supporting experimental results, and complete characterization data for all new compounds. See DOI: https://doi.org/10.1039/d5sc10040b.CCDC 2403829 contains the supplementary crystallographic data for this paper.22
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2022YFA1503703), the National Natural Science Foundation of China (No. 22325106 and 22471204), the Fundamental and Interdisciplinary Disciplines Breakthrough Plan of the Ministry of Education of China (No. JYB2025XDXM402) and the Taikang Center for Life and Medical Sciences of Wuhan University. We thank Prof. W.-B. Liu (WHU) for sharing the instruments, and Mr W. Yuan and Prof. Z. Li (WHU) for their kind assistance with the fluorescence quantum yield measurements.Notes and references
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom., 1994, 19, 17–39 CrossRef.
- (a) J.-W. Han, J.-X. Chen, X. Li, X.-S. Peng and H. N. C. Wong, Synlett, 2013, 24, 2188–2198 CrossRef CAS; (b) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC; (c) G. Wu, Y. Liu, Z. Yang, T. Jiang, N. Katakam, H. Rouh, L. Ma, Y. Tang, S. Ahmed, A. U. Rahman, H. Huang, D. Unruh and G. Li, Natl. Sci. Rev., 2019, 7, 588–599 CrossRef PubMed; (d) Y. Liu, G. Wu, Z. Yang, H. Rouh, N. Katakam, S. Ahmed, D. Unruh, Z. Cui, H. Lischka and G. Li, Sci. China:Chem., 2020, 63, 692–698 CrossRef CAS; (e) M. Tang and X. Yang, Eur. J. Org Chem., 2023, 26, e202300738 CrossRef CAS; (f) J. Feng, L.-L. Xi and R.-R. Liu, Chem.–Eur. J., 2025, 31, e02247 CrossRef CAS PubMed; (g) J. Feng, L.-L. Xi and R.-R. Liu, Trends Chem., 2025, 7, 567–570 CrossRef CAS; (h) K. Zhu, D. R. Spring, B.-F. Shi and F. Zhang, Chem. Soc. Rev., 2025, 54, 10856–10879 RSC; (i) W. Qin and G. Cera, Chem. Rec., 2025, 25, e202400237 CrossRef CAS PubMed.
- (a) M. Durmaz, S. Alpaydin, A. Sirit and M. Yilmaz, Tetrahedron: Asymmetry, 2007, 18, 900–905 CrossRef CAS; (b) S. Shirakawa, T. Kimura, S.-i. Murata and S. Shimizu, J. Org. Chem., 2009, 74, 1288–1296 CrossRef CAS PubMed; (c) S.-Y. Li, Y.-W. Xu, J.-M. Liu and C.-Y. Su, Int. J. Mol. Sci., 2011, 12, 429–455 CrossRef CAS PubMed; (d) P. Nandi, A. Solovyov, A. Okrut and A. Katz, ACS Catal., 2014, 4, 2492–2495 CrossRef CAS; (e) G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS PubMed; (f) P. Lhoták, Org. Biomol. Chem., 2022, 20, 7377–7390 RSC.
- J. K. Browne, M. A. McKervey, M. Pitarch, J. A. Russell and J. S. Millership, Tetrahedron Lett., 1998, 39, 1787–1790 CrossRef CAS.
- K. Ishibashi, H. Tsue, H. Takahashi and R. Tamura, Tetrahedron: Asymmetry, 2009, 20, 375–380 CrossRef CAS.
- (a) S. Tong, Acc. Chem. Res., 2025, 58, 3574–3591 CrossRef CAS PubMed; (b) S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed; (c) Y.-F. Jiang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2024, 15, 12517–12522 RSC; (d) X.-C. Li, Y. Cheng, X.-D. Wang, S. Tong and M.-X. Wang, Chem. Sci., 2024, 15, 3610–3615 RSC; (e) Q.-L. Lu, X.-D. Wang, S. Tong, J. Zhu and M.-X. Wang, ACS Catal., 2024, 14, 5140–5146 CrossRef CAS.
- Y.-Z. Zhang, M.-M. Xu, X.-G. Si, J.-L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858–22864 CrossRef CAS PubMed.
- X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2023, 14, 827–832 RSC.
- (a) G. Giovanardi, G. Scarica, V. Pirovano, A. Secchi and G. Cera, Org. Biomol. Chem., 2023, 21, 4072–4083 RSC; (b) M. Goudarzi, A. Alagiyawanna, A. Serafino, S. Rizzato, F. Bertocchi, F. Terenziani, S. Santoro, W. Qin, G. Cera and V. Pirovano, Chem. Commun., 2025, 61, 16380–16383 RSC.
- X.-Y. Zhang, D. Zhu, R.-F. Cao, Y.-X. Huo, T.-M. Ding and Z.-M. Chen, Nat. Commun., 2024, 15, 9929 CrossRef CAS PubMed.
- (a) M. Yuan, W. Xie, S. Yu, T. Liu and X. Yang, Nat. Commun., 2025, 16, 3943 CrossRef CAS PubMed; (b) X. Gong, L. Zhang, X. Liu, M. Shi, M. Zhang, T. Yao, Q. Yan and J. Nan, Org. Lett., 2025, 27, 12991–12996 CrossRef CAS PubMed; (c) M. Yuan, W. Xie and X. Yang, Chin. Chem. Lett., 2025 DOI:10.1016/j.cclet.2025.112157.
- Y. Yao, F. Wang, X.-X. Li, R. Mi and X. Li, Angew. Chem., Int. Ed., 2025, 64, e202520283 CrossRef CAS PubMed.
- V. Dočekal, L. Lóška, A. Kurčina, I. Císařová and J. Veselý, Nat. Commun., 2025, 16, 4443 CrossRef PubMed.
- L. Peng, Y. Chang, S. Tong, W. Qin, G. Cera and H. Yan, Angew. Chem., Int. Ed., 2025, 64, e202518659 CrossRef CAS PubMed.
- (a) Y.-K. Jiang, Y.-L. Tian, J. Feng, H. Zhang, L. Wang, W.-A. Yang, X.-D. Xu and R.-R. Liu, Angew. Chem., Int. Ed., 2024, 63, e202407752 CrossRef CAS PubMed; (b) S. Yu, M. Yuan, W. Xie, Z. Ye, T. Qin, N. Yu and X. Yang, Angew. Chem., Int. Ed., 2024, 63, e202410628 CrossRef CAS PubMed; (c) X.-Y. Fan, Q.-X. Li, T.-Y. Zhai, S.-F. Ni, G. Wu, L.-W. Ye and B. Zhou, ACS Catal., 2026, 16, 818–830 CrossRef CAS.
- (a) T. Li, Y. Zhang, C. Du, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2024, 15, 7673 CrossRef CAS PubMed; (b) P.-F. Qian, G. Zhou, J.-H. Hu, B.-J. Wang, A.-L. Jiang, T. Zhou, W.-K. Yuan, Q.-J. Yao, J.-H. Chen, K.-X. Kong and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202412459 CrossRef CAS PubMed; (c) L. Zhang, C. Yang, X. Wang, T. Yang, D. Yang, Y. Dou and J.-L. Niu, Green Chem., 2024, 26, 10232–10239 RSC.
- (a) R. Li, F. Liu and G. Dong, Org. Chem. Front., 2018, 5, 3108–3112 RSC; (b) H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581–585 CrossRef CAS PubMed; (c) Z.-S. Liu, Y. Hua, Q. Gao, Y. Ma, H. Tang, Y. Shang, H.-G. Cheng and Q. Zhou, Nat. Catal., 2020, 3, 727–733 CrossRef CAS; (d) Q. Feng, X. Ma, W. Bao, S.-J. Li, Y. Lan and Q. Song, CCS Chem., 2021, 3, 377–387 CrossRef CAS; (e) Y. Hua, Z.-S. Liu, P.-P. Xie, B. Ding, H.-G. Cheng, X. Hong and Q. Zhou, Angew. Chem., Int. Ed., 2021, 60, 12824–12828 CrossRef CAS PubMed; (f) Z.-S. Liu, P.-P. Xie, Y. Hua, C. Wu, Y. Ma, J. Chen, H.-G. Cheng, X. Hong and Q. Zhou, Chem, 2021, 7, 1917–1932 CrossRef CAS; (g) B. Ding, Q. Xue, H. Wei, J. Chen, Z.-S. Liu, H.-G. Cheng, H. Cong, J. Tang and Q. Zhou, Chem. Sci., 2024, 15, 7975–7981 RSC; (h) Q. Tian, J. Ge, Y. Liu, X. Wu, Z. Li and G. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202409366 CrossRef CAS PubMed; (i) J. Ye, Y. You, D. Xu, S. Deng, L. Qi, C. Liu, C.-C. Liu, H. Cong, P. Wang, H.-G. Cheng and Q. Zhou, CCS Chem., 2025, 7, 3025–3035 CrossRef CAS; (j) Q. Tian, J. Ge, Y. Liu, X. Wu, Z. Li and G. Cheng, Org. Lett., 2025, 27, 121–128 CrossRef CAS PubMed; (k) Z. Cui, H.-G. Cheng and Q. Zhou, ChemCatChem, 2025, 17, e202500560 CrossRef CAS; (l) L. Li, J. Ye, L. Zhao, L. Zhou, S. Deng, D. Wang, Y. Zhang, H. Cong, Q. Zhou and H.-G. Cheng, J. Am. Chem. Soc., 2025, 147, 39721–39731 CrossRef CAS PubMed.
- (a) M. Catellani, F. Frignani and A. Rangoni, Angew Chem. Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS; (b) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed; (c) N. Della Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed; (d) H.-G. Cheng, S. Chen, R. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2019, 58, 5832–5844 CrossRef CAS PubMed; (e) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed; (f) K. Zhao, L. Ding and Z. Gu, Synlett, 2019, 30, 129–140 CrossRef CAS; (g) S. Dong and X. Luan, Chin. J. Chem., 2021, 39, 1690–1705 CrossRef CAS.
- Q. Gao, C. Wu, S. Deng, L. Li, Z.-S. Liu, Y. Hua, J. Ye, C. Liu, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, J. Am. Chem. Soc., 2021, 143, 7253–7260 CrossRef CAS PubMed.
- (a) Y. An, X.-Y. Zhang, Y.-N. Ding, Y. Li, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2022, 24, 7294–7299 CrossRef CAS PubMed; (b) J. Ye, L. Li, Y. You, C. Jiao, Z. Cui, Y. Zhang, S. Jia, H. Cong, S. Liu, H.-G. Cheng and Q. Zhou, JACS Au, 2023, 3, 384–390 CrossRef CAS PubMed.
- C.-J. Wang, Z.-Y. Xue, G. Liang and Z. Lu, Chem. Commun., 2009, 2905–2907 RSC.
- CCDC 2403829: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2lpcwk.
| This journal is © The Royal Society of Chemistry 2026 |