
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiscale insights into sodium storage in hard carbon from operando small- and wide-angle neutron scattering measurements
Yoshihiko
Umemoto
 *ab,
Kazuki
Ohishi
*c,
Daisuke
Igarashi
d,
Ryoichi
Tatara
e,
Che-an
Lin
f,
Kosuke
Nakamoto
d,
Yukihiko
Kawamura
c,
Kosuke
Hiroi
g,
Shin-ichi
Takata
g,
Yusuke
Nambu
hi,
Yoshitaka
Tateyama
f and
Shinichi
Komaba
d
*ab,
Kazuki
Ohishi
*c,
Daisuke
Igarashi
d,
Ryoichi
Tatara
e,
Che-an
Lin
f,
Kosuke
Nakamoto
d,
Yukihiko
Kawamura
c,
Kosuke
Hiroi
g,
Shin-ichi
Takata
g,
Yusuke
Nambu
hi,
Yoshitaka
Tateyama
f and
Shinichi
Komaba
d
aInstitute for Materials Research, Tohoku University, Miyagi 980-8577, Japan. E-mail: yoshihiko.umemoto.t6@alumni.tohoku.ac.jp; Fax: +81 (0)22 215 2327; Tel: +81 (0)22 215 2327
bNeutron Technologies Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
cNeutron Science and Technology Center, Comprehensive Research Organization for Science and Society (CROSS), Ibaraki 319-1106, Japan. E-mail: k_ohishi@cross.or.jp; Fax: +81 (0)29 219 5311; Tel: +81 (0)29 219 5310
dDepartment of Applied Chemistry, Tokyo University of Science, Tokyo 162-8601, Japan
eDepartment of Chemistry and Life Science, Yokohama National University, Kanagawa 240-8501, Japan
fSchool of Materials and Chemical Technology, Institute of Science Tokyo, Kanagawa, 226-8501, Japan
gJ-PARC Center, Japan Atomic Energy Agency (JAEA), Ibaraki 319-1195, Japan
hInstitute for Integrated Radiation and Nuclear Science, Kyoto University, Osaka 590-0494, Japan
iFOREST, Japan Science and Technology Agency, Saitama 332-0012, Japan
First published on 25th February 2026
Abstract
Understanding the sodium (Na) storage mechanism in hard carbon (HC) is critical for the development of high-performance Na-ion batteries, which are a promising alternative to lithium-ion batteries owing to the abundance and low cost of Na. In this study, we investigate the Na storage mechanism in commercial HC using operando small- and wide-angle neutron scattering (SANS and WANS). The experiments cover a wide range of scattering vectors, enabling the simultaneous observation of the structural evolution of nanoporous regions and graphene layers during electrochemical charging. Both SANS and WANS analyses reveal that Na storage proceeds sequentially via initial surface adsorption, subsequent insertion into graphene layers, and final filling of nanopores. These results demonstrate the effectiveness of simultaneous operando SANS and WANS measurements for elucidating the complex, multiscale mechanisms of Na storage in HC.
1 Introduction
High-performance batteries reduce wasted energy. Despite the high density and long cycle life of lithium-ion batteries (LIBs) concerns about limited lithium supply and environmental impact have driven interest in alternative energy storage technologies.1 Sodium (Na), being more abundant and widely distributed, makes Na-ion batteries (NIBs) a promising alternative to LIBs, providing greater cost-effectiveness in the long term.2,3 In LIBs, graphite is commonly used as the anode because of its efficient lithium-ion insertion.4 In contrast, the lower energetic stability of Na binary graphite insertion formation potentials challenges Na insertion into graphite.5,6 To enable efficient Na storage, alternative carbon materials with disordered structures are required. In the early 21st century, hard carbon (HC), a non-graphitizable carbon with a disordered structure, was identified as a promising anode material because of its ability to store Na ions via insertion and adsorption mechanisms.7,8 Advances in synthesis methods, including the pyrolysis of biomass and polymer precursors, have led to the development of high-capacity HC materials with improved cycle stability and rate performance.9–11 Recent studies have focused on optimizing the pore structure, surface chemistry, and electrolyte compatibility to further enhance the performance of HC anodes.12–16In 2021, Kamiyama et al. reported that HC materials synthesized using the MgO-template method exhibit a significant reversible capacity and high initial coulombic efficiency.17 MgO-template synthesis is a widely used approach for fabricating porous carbon and nanostructured materials, utilizing MgO as a sacrificial template to control the morphology, porosity, and structure of the final product. In addition, ZnO and CaCO3 have been investigated as nanopore templates, with ZnO proving to be the most effective.18–20 These findings highlight the importance of characterizing nanopore structures and monitoring Na storage evolution during the charging/discharging processes.
Na storage in HC proceeds via several mechanisms, including insertion between graphene sheets, adsorption at reactive surface sites, and nanopore filling.7,21 The charge/discharge profile of HC typically consists of two distinct regions: a sloping region at potentials above 0.1 V and a plateau region at potentials below 0.1 V, where significant capacity is gained at an almost constant potential. Many studies have attempted to elucidate the correlation between the Na storage mechanism and the charge/discharge profile. However, the structural complexity of HC makes it challenging to assign these processes definitively.7,14,21,22
Although structural analysis in reciprocal space using X-rays and neutrons is well suited for investigating HC materials, a key difficulty arises from their multiscale structure.23–29 For example, small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS) can be used to determine the average diameter of nanopores at the nanometer scale, whereas graphene layer structures, corresponding to the high-angle region, exist at the angstrom scale. Scattering instruments capable of simultaneously covering both small- and high-angle regions with a wide solid angle remain scarce, and operando experimental cells optimized for effectively using such wide-angle coverage have yet to be developed.
In this study, we present a SANS and wide-angle neutron scattering (WANS) investigation of the Na storage process in commercial HC using a developed operando cell.30
2 Experimental
2.1 Sample preparation
HC (95 wt%, Carbotron P(J), Kureha) was mixed with a Na polyacrylate binder (5 wt%, Kishida Chemical) and water to create a slurry before being cast on a copper (Cu) current collector to obtain a thickness of 0.05 mm and cover a circular diameter of 10 mm to match the neutron beam aperture. The Na half-cell was assembled in an argon-filled glovebox (H2O < 0.5 ppm). The cell consisted of a 0.1 mm-thick rolled Na metal (Kanto Chemical) as the counter electrode, a quartz fiber filter (QR-100, ADVANTEC) as the separator, and a 1 mL of a 1 mol per kg NaPF6 solution (Kishida Chemical) as the electrolyte. The electrolyte solvent was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture deuterium exchanged ethylene carbonate and diethyl carbonate (FUJIFILM Wako Pure Chemical).
:1 (v/v) mixture deuterium exchanged ethylene carbonate and diethyl carbonate (FUJIFILM Wako Pure Chemical).
2.2 Operando SANS and WANS experiment
We employed TAIKAN,31 a SANS and WANS instrument at BL15 of the Materials and Life Science Experimental Facility (MLF) at the Japan Proton Accelerator Research Complex (J-PARC), to investigate the Na storage mechanism in HC over a wide q range (0.05–50 nm−1). We have employed muon and neutron based techniques from multiple perspectives to comprehensively understand the structures and ion dynamics of both the battery anodes and cathodes.32–35 We modified the operando muon cell developed in our previous study to develop an operando SANS cell specifically optimized for use in TAIKAN.30 We used our newly developed operando cell to observe the structural changes in HC during charging and to correlate them with the Na storage process. The details of the operando cell are described in the SI.The operando charge sequences were performed using a Biologic SP-300 within a potential window of 0.01–2.5 V vs. Na+/Na at a rate of C/50 (the current was calculated assuming a practical capacity of 250 mAh g−1). Fig. 1(a) shows the initial charge curve for HC in a half-cell of the experiment. The operando SANS and WANS measurements were performed over 60 h. We segmented the operando data into 3 h intervals and converted them to absolute intensity units to provide an overview of the structural changes in HC during charging. The data were sliced into 3 h segments using Utsusemi software,36,37 which was developed for analyzing and visualizing neutron scattering data at MLF. The absolute intensity was obtained using a glassy carbon standard sample. The lower axis of Fig. 1(a) corresponds directly to these time slices. The charging capacity is shown on the upper axis of Fig. 1(a), while the potential is indicated on the left axis. This representation allows each 3-hour slice to be associated with an approximate state of charge in terms of both capacity and potential.
 | ||
| Fig. 1 (a) Electrochemical initial charge curve. (b) Reference SANS and WANS profile, and (c) pre-charged measurement profile. Inset of (b) and (c) show the high-angle Bragg peak region. | ||
Density functional theory (DFT) calculations were performed using the generalized gradient approximation with Perdew–Burke–Ernzerhof functional as the exchange–correlation38,39 and a pseudopotential based on the projector augmented-wave method implemented in the Vienna ab initio simulation package (VASP).40–43 The DFT-D3 dispersion correction functional implemented in VASP was used.44 The cutoff energy value was 520 eV, and a k-point mesh of 2 × 2 × 2 was used. All computations were converged to 10−5 eV by energy and to 10−2 eV Å−1 by force. The atomic models were visualized through VESTA.45
3 Results
3.1 Profiles
Fig. 1(b) and (c) show typical SANS and WANS profiles. A hump feature, which can be attributed to the nanopore structure in HC, appears at q ∼ 1 nm−1, which is consistent with the ex situ results on the same HC.34 The WANS region exhibits a profile similar to that in Fig. 1(b). The broad peaks at q ∼ 15 nm−1 contain contributions from both the electrolyte and the graphene layer components in HC. Fig. 1(b) presents the reference profile, which includes a Cu current collector plate, a Na metal electrode, a separator, and the electrolyte (without HC). As shown in the inset of Fig. 1(b), broad peaks originating from the electrolyte, along with sharp Bragg peaks from the Cu current collector plate and Na metal electrode, are observed in the WANS region. Fig. 1(c) shows the pre-charged measurement profile (with HC). A systematic evolution of the SANS and WANS profile was observed during charging. To quantitatively assess these changes, we conducted the following analysis.3.2 WANS region analysis
Fig. 2(a) shows the reference profile, the pre-charged measurement profile, and their difference profile. The typical layer distance of graphite is 0.335 nm,46 which corresponds to a peak at ∼18.8 nm−1 in reciprocal space. As shown in the difference profile in Fig. 2(a), a broad graphene layer peak appears at approximately 17.5 nm−1, reflecting the low crystallinity of HC. Fig. 2(c) presents the difference profile between the reference profile and the profile during charging. The peak position in the difference profile shifts toward lower q and becomes broader. | ||
| Fig. 2 (a) WANS profiles of reference, pre-charged, and their difference. (b) Fitting result of pre-charged measurement profile (solid orange line). The dotted line corresponds to the electrolyte component, the solid green line corresponds to the HC component, the solid black line corresponds to the Na metal, and the dashed line corresponds to the background component. (c) Evolution of the difference between operando and reference profiles during the charging process. Initial charge time dependence of analysis parameter: (d) peak center, (e) FWHM, (f) intensity, and (g) background of the fitting parameters. The white circles represent the fitting result for the pre-charged state, and the black circles represent the charging state. The solid lines are guides for the eyes. | ||
Because of its lower intensity than the broad electrolyte peak around 15 nm−1 and the increasing constant background, the evolution of the graphene layer peak in HC remains difficult to resolve. To quantitatively evaluate subtle changes in the graphene layer structure of HC and to elucidate the Na storage behavior during charging, we analyzed the WANS profiles using eqn (1). A representative result of the analysis is presented in Fig. 2(b). In this equation, I, w, and c denote the intensity, full width at half maximum (FWHM), and peak center of the Lorentzian function, respectively.
 | (1) |
The first term in eqn (1) represents the broad peak originating from the electrolyte. The second term corresponds to the Bragg peak of Na metal at q ∼ 21 nm−1. The parameters for these two terms were fixed using the parameters obtained by fitting the reference data. The third term corresponds to the graphene layer peak of HC, which is treated as a free parameter in the analysis of the operando profiles. The fourth and fifth terms account for the background, modeled as a linear function and a constant term, respectively. Because the background profile could be adjusted by adding or subtracting constants, the constant background term in the fifth component was attributed the variations in the incoherent scattering contribution and was treated as a free parameter in all profiles. The corresponding results of the third term and constant background term are shown in Fig. 2(d)–(g). In these figures, the white circles represent the fitting results for the pre-charged state, and black circles represent the charging state. The peak position remains nearly unchanged for the first 12 h, then shifts significantly toward lower q during the latter half of the slope region, and finally saturates in the plateau region. The full width at FWHM also remains nearly constant for the first 12 h, begins to increase from the later part of the slope region, and saturates in the plateau region. These trends qualitatively reproduce the behavior of the difference profile shown in Fig. 2(c). These results indicate that, in the slope region, the graphene layer spacing expands and the structure becomes more disordered, whereas in the plateau region, structural changes are relatively suppressed. The HC peak intensity remains almost unchanged throughout the charging process.
3.3 SANS region analysis
As shown in Fig. 3(a), the small-angle scattering profile exhibits a characteristic hump, the intensity of which decreases during charging. To quantitatively analyze the behavior of this hump, the following analysis was performed. The hump observed in the small-angle region, which corresponds to the nanoporous structure of HC, was primarily analyzed using either the Debye–Bueche model47,48 or the Teubner–Strey model.49 The Debye–Bueche model is a classical scattering approach that describes systems containing randomly distributed inhomogeneities or density fluctuations, such as porous materials or disordered two-phase systems.47,48 It assumes an exponential correlation of density variations and is typically applied to structures that lack long-range order, exhibiting little or no structural periodicity. This model has been used in to both SANS and SAXS measurements to evaluate the nanopore size in HC subjected to different heat treatment conditions.7,14,29 The Teubner–Strey model, originally proposed by Teubner and Strey in 1987,49 is a theoretical framework used to describe structures in which two phases are mixed with nanoscale periodicity, such as microphase-separated or bicontinuous systems.50 This model is applicable to systems with partially periodic structures and can reproduce characteristic features in the scattering profile, such as broad peaks and shoulders.26,51,52 Recent operando SANS studies of HCs52 have employed the Teubner–Strey model for structural analysis. In this study, we initially attempted to apply the same model to our data. However, the fitting resulted in an unphysically large pore–pore distance that approaches infinity, indicating that the assumption of spatially correlated pores is not valid in the present case. We considered the pores to be uncorrelated, corresponding to a fully random distribution without periodicity. Note that the HC used in our study was obtained from a different manufacturer than that used in the previous study.52 | ||
| Fig. 3 (a) Change in the hump region of the SANS profile during initial charging. The purple solid triangles indicate the pre-charged state, and the black filled squares indicate the overcharged state. (b) Fitting result of the pre-charged SANS profile (solid orange line). The dotted line corresponds to the Porod component, the solid green line corresponds to the hump component, and the dashed line corresponds to the background. Initial charge time dependence of analysis parameter: (c) intensity, (d) exponential part of the Porod region, (e) intensity of nanopore region Inanopore, and (f) background with charging. The white circles represent the fitting result for the pre-charged state, and the black circles represent the charging state. The solid lines are guides for the eyes. | ||
Consequently, the analytical expression used for the small-angle region is the Debye–Bueche model, which is given in eqn (2).
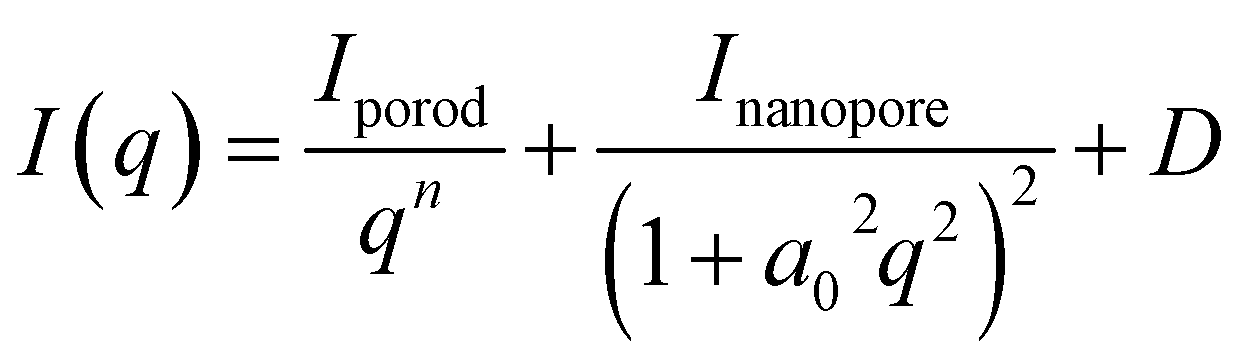 | (2) |
 . Assuming that the pore size remains constant during charging, the a0 parameter was determined from the pre-charged measurement profile as a0 = 0.3161(5), and this value was subsequently used in the profile analysis during charging. The resulting average pore radius, R = 0.9995(15) nm, is reasonable compared with typical nanopore sizes reported for HC. The typical ionic radius of Na+ is approximately 0.1 nm. Assuming spherical nanopores, the pore volume is roughly 103 times larger than that of a single Na+ ion. This large difference in length and volume scales indicates that Na+ insertion into nanopores does not cause significant distortion or collapse of the carbon framework. Based on these results, we consider it reasonable to assume that the nanopore size remains essentially constant during the charging process. Moreover, we confirmed that allowing a0 to vary does not change the overall trend of Inanopore as shown in SI. The third term in eqn (2) represents the constant background and is treated as a free parameter in all profile fittings to account for the variations in incoherent scattering during charging, similar to the analysis in the WANS region.
. Assuming that the pore size remains constant during charging, the a0 parameter was determined from the pre-charged measurement profile as a0 = 0.3161(5), and this value was subsequently used in the profile analysis during charging. The resulting average pore radius, R = 0.9995(15) nm, is reasonable compared with typical nanopore sizes reported for HC. The typical ionic radius of Na+ is approximately 0.1 nm. Assuming spherical nanopores, the pore volume is roughly 103 times larger than that of a single Na+ ion. This large difference in length and volume scales indicates that Na+ insertion into nanopores does not cause significant distortion or collapse of the carbon framework. Based on these results, we consider it reasonable to assume that the nanopore size remains essentially constant during the charging process. Moreover, we confirmed that allowing a0 to vary does not change the overall trend of Inanopore as shown in SI. The third term in eqn (2) represents the constant background and is treated as a free parameter in all profile fittings to account for the variations in incoherent scattering during charging, similar to the analysis in the WANS region.
First, the pre-charged SANS profile was fitted. The results are presented in Fig. 3(b). As illustrated in Fig. 3(a), the slope in the Porod region remains nearly unchanged between the pre-charged and over-charged states, indicating that no significant structural changes occur at length scales larger than 10 nm. This is also evident from the fact that the intensity and exponent in the Porod region (Fig. 3(c) and (d)) remain virtually unchanged throughout the charging process. The evolution of Inanopore as a function of the charging time is presented in Fig. 3(e). The value of Inanopore remains nearly constant for approximately the first 12 h of charging but begins to decrease during the latter half of the sloping region. The variation in the constant background with charging time is shown in Fig. 3(f). Although the background tends to increase during charging, the magnitude of this change is small compared with the overall change in Inanopore.
4 Discussion
The center position of the HC peak, cHC, corresponds to the averaged graphene layer distance, L. As shown in Fig. 4(a), the graphene layer distance L, increases with charging, probably reflecting charge-induced Na insertion into the graphene layers. The HC peak width, wHC (Fig. 2(e)) also increases with charging until approximately 30 h, coinciding with the onset of the plateau region. This broadening indicates that Na insertion broadened the distribution of the graphene layer distances in HC. | ||
| Fig. 4 (a) Graphene layer distance L (left axis) and volume fraction of Na+ inserted layer V (right axis) as a function of charging time. The white open circles represent the pre-charged measurement profile, and the black filled circles represent the charged profile. The blue dashed line indicates the approximate boundary between the region in which Na+ insertion graphene layers is dominant and the region in which Na+ filling into nanopores is dominant. The thick purple line serves as a guide for the eye to illustrate the increase in the graphene layer distance and Na insertion amount. (b) Change in the graphene layer distance upon Na+ insertion into a graphene layer, as calculated theoretically. The schematic on the right side of the graph illustrates the insertion pattern into the graphene layer for representative Na amounts. To clarify the correspondence between the vertical axis ranges of (a) and (b), the light blue shaded background indicates the same vertical axis range as in (a). (c) Normalized intensity Inanopore(t)/Inanopore(0) of the SANS profile during charging. The white open circles represent the pre-charged measurement profile, and the black filled circles represent the charged profile. The blue dashed line indicates the approximate boundary between the region in which Na+ insertion into graphene layers is dominant and the region in which Na+ filling into nanopores is dominant. (d)–(f) Schematic diagram of the SLD for phase ϕ1 and ϕ2. The orange arrows indicate the ΔSLD, which corresponds to the Inanopore. (d) Pre-charged sate, (e) Na+ insertion into the carbon framework, and (f) Na+ filling into nanopore region. | ||
To quantify the total volume of Na ions inserted into HC, we consider the simplest model. We define Lini as the initial empty graphene layer distance of HC and LNa as the graphene layer distance upon the insertion of a single Na ion between the graphene layers. Assuming that all graphene layers contain either none or one layer of Na or none at all, the relationship between the experimentally observed graphene layer distances, L and Lini, and the volume fraction of the graphene layer containing Na is described using eqn (3).
| LNaV + Lini(1 − V) = L | (3) |
The absolute value of V depends on LNa. Because this experiment does not allow for the direct determination of LNa, we estimate it based on the following discussion. According to recent research, the graphene layer distance of Li-inserted graphite is LLi = 0.3709 nm, whereas that of K-inserted graphite is LK = 0.535 nm.19,20 By assuming that the graphene layer spacing for Na+ insertion falls between these values, LNa = 0.453 nm is expected. Then, the expansion from the pure graphene layer distance (0.335 nm) is approximately 0.118 nm: thus LNa = Lini + 0.118. Based on the aforementioned previous studies on graphene layer distances, we assume that the spacing of the graphene layer in the HC sample increases by 0.118 nm upon Na insertion. Accordingly, we adopted an elongation value of 0.118 nm to evaluate the maximum possible degree of insertion. Consequently, V is given by eqn (4).
 | (4) |
As evident from eqn (4), L and V exhibit a proportional relationship; therefore, the volume fraction V is shown on the right axis in Fig. 4(a). The volume fraction V increases rapidly from the middle of the slope region to the onset of the plateau region (approximately 40 h after the start of charging) and subsequently saturates. This behavior indicates that Na insertion into graphene-like layers is most active during the latter half of the slope region, whereas further insertion becomes much less significant in the plateau region.
In the fully charged state, the interlayer distance of the graphene-like layers and the corresponding volume fraction of Na+-inserted layers are approximately 0.389(2) nm and V ≈ 28%, respectively. This result implies that a substantial fraction of the graphene-like layers remains unoccupied by Na+ even at full charge. It should be noted that, because the reference state assumes complete Na insertion into ideal graphite layers, the absolute number of vacant graphene-like layers cannot be uniquely determined for structurally disordered materials such as HC. Nevertheless, although the exact number of empty layers in HC is difficult to quantify, adopting a fixed reference state enables a consistent comparison of Na filling susceptibility among different HC materials. By evaluating the relative interlayer expansion ratio defined in eqn (4), rather than the absolute interlayer distance, one can quantitatively assess not only whether Na+ inserts more readily into one HC material than into another, but also how closely the graphene like layers structure approaches an ideal graphite layered structure.
Fig. 4(b) shows the theoretically calculated average graphite crystal layer distances for NaC6 formed by Na+ insertion into graphite layers. The horizontal axis (NaxC240) represents the Na insertion concentration (x), while the vertical axis denotes the average graphite crystal layer distance. In Li-ion batteries, the maximum Li+ insertion into graphite anodes at the fully charged state is well established as LiC6 (Li40C240). By analogy, the maximum Na+ insertion into HC is assumed to correspond to NaC6 (Na40C240). In the calculation, four graphite layers were considered, and Na+ ions were assumed to be inserted into the next interlayer only after the preceding layer became fully occupied. A discontinuous increase in the average graphite layer distance occurs when Na+ is first inserted into a given layer, whereas continuous expansion is observed as Na+ progressively fills the same layer. This behavior indicates that the apparent linearity of graphene layer expansion is governed by the specific filling sequence of individual layers. Experimentally, as shown in Fig. 4(a), the graphene-like layer distance increases steadily until approximately 30 h after the start of charging and reaches about 0.39 nm. According to the DFT calculations with graphite crystal layers, this interlayer distance corresponds to x = 18, which is equivalent to approximately 45% of the fully sodiated state, Na40C240, in which all graphite interlayers are completely filled with Na+. In contrast, the experimentally estimated occupation ratio of Na+ within the graphene-like layers is only about 28%. Experimentally, Na+ is accommodated not only within the interlayer galleries of graphene-like layers but also at basal planes, edge sites, and within nanopore regions. Thus, the limited contribution of the graphene-like layers (∼28%) provides a reasonable explanation for the observed saturation behavior and is qualitatively consistent with the simulations. The remaining discrepancy between experiment and calculation implies that the graphene-like layers in HC exhibit a higher degree of structural randomness than those in graphite crystal. To enable a direct comparison between calculation and experiment, the interlayer distance of the pristine state in the calculation model was set to the experimentally determined value rather than the nominal graphite spacing. As the calculated interlayer distance reaches the experimentally observed saturation value of approximately 0.39 nm at (x = 18), the results indicate that about 45% of the chemically allowable Na+ is accommodated within only approximately 28% of the available interlayer volume in HC. Although it remains experimentally unclear whether excess free space persists within the graphene-like layers, the chemical permissibility of further Na+ insertion suggests that the interlayer Na+ storage capacity could be enhanced by optimizing the disordered structure of HC. It should be noted, however, that the relationship between structural disorder and Na+ insertion capacity is complex. Interlayer Na+ insertion is governed not only by the degree of disorder but also by factors such as the density and edge-termination states of graphene like layers domains. Furthermore, whether an increase in the amount of intercalation directly leads to an improvement in the overall capacity of HC is a separate issue. As discussed below, the present study and previous research52 suggest that intercalation and pore-filling proceed sequentially. While an increase in one does not fundamentally hinder the other, it remains an open question whether these two mechanisms act synergistically or exhibit a trade-off relationship during the HC manufacturing process; thus, further investigation is required.
The intensity of the SANS profile, Inanopore, has the following relationship in the Debye–Bueche model:
| Inanopore ∝ ϕ1ϕ2(ρ1 − ρ2)2, | (5) |
| ΔSLD = ρ1 − ρ2, | (6) |
By considering both the SANS and WANS results, we gain detailed insights into the Na+ storage process in HC. The WANS measurements demonstrate that the volume fraction of Na+ storage V increases in the slope region but saturates in the plateau region (approximately 30 h after the initiation of charging), suggesting that Na+ insertion into the graphene layers is no longer dominant in the plateau region. Therefore, to account for the constant decrease of Inanopore(t)/Inanopore(0) in the SANS region, we conclude that Na storage in the nanopore region becomes dominant during the plateau phase. The Debye–Bueche model represents a random two-phase system. Therefore, we consider it in three distinct regions: pre-charged, slope, and plateau. These three regions correspond to the case illustrated in Fig. 4(d)–(f), respectively. Although the mechanisms causing the decrease in ΔSLD differ, irrespective of whether via Na+ insertion into graphene layers or nanopores, the rate of decrease in Inanopore(t)/Inanopore(0) remains effectively unchanged, because the differences are too subtle to be detected under the present experimental conditions. Furthermore, in both Fig. 4(a) and (c), the sharp increase in Na insertion into the graphene layer regions and the corresponding rapid decrease in Inanopore in the overcharge regime suggest that Na is effectively “pushed” into the graphene layers under overcharge conditions. To further investigate the reversibility of these structural changes, operando SANS and WANS measurements during repeated charging and discharging cycles are required. Such studies will help clarify the reversibility of overcharging-induced structural modifications and identify the spatial scales of any irreversible changes.
As previously mentioned, a flat region is observed at the beginning of the slope region. In this experiment, the first region indicated the first charging process; therefore, when Na is introduced for the first time into the HC, it is expected to be trapped at the HC edges and defect sites, consistent with previous research. After this process is completed, the insertion of Na+ into the graphene layers begins.
Finally, our results differ from previous operando SANS experiments on HC,52 which reported a distinct trend in the decrease in ΔSLD2 between the slope and plateau regions. In the previous study, the authors calculated ΔSLD by accounting for expansion of the volume fraction of HC during the charging process, as inferred from the reference film results.55 Detailed analyses and further discussion of the differences from the previous study52 are provided in the SI.
5 Conclusions
This study employed operando SANS and WANS using a custom-designed cell compatible with the TAIKAN instrument to investigate the Na storage mechanism in HC anodes. The wide-q-range setup enabled the simultaneous observation of the structural changes in both nanopores and graphene layers during charging.The WANS measurements quantitatively captured the structural changes in HC associated with Na insertion into the graphene layers, revealing that graphene layer expansion and structural disorder predominantly occurred in the slope region and became saturated in the plateau region. The measured graphene layer distances showed good agreement with the theoretical calculations, highlighting the complementarity between the experimental and theoretical results. These findings highlight the importance of high-q structural analysis of amorphous materials such as HC.
For the SANS observations, a new interpretation of the intensity changes was proposed under the realistic assumption that the HC volume remains constant and that the electrolyte does not penetrate the HC structure. The results demonstrate that the SANS intensity decreases during charging, which can be primarily attributed to Na insertion into graphene layers in the slope region and into nanopores in the plateau region.
By integrating the SANS and WANS measurements, clear evidence was obtained for a continuous Na storage process in which nanopores are filled after the initial insertion into the graphene layers. Overall, this study demonstrates the effectiveness of neutron scattering over a broad q range in elucidating complex Na storage mechanisms in disordered carbon materials.
Author contributions
Conceptualization: Kazuki Ohishi, Daisuke Igarashi, Ryoichi Tatara, Yoshitaka Tateyama, Shinichi Komaba; investigation: Yoshihiko Umemoto, Kazuki Ohishi, Daisuke Igarashi, Ryoichi Tatara, Che-an Lin, Kosuke Nakamoto, Yukihiko Kawamura, Kosuke Hiroi, Shin-ichi Takata; supervision: Kazuki Ohishi, Yusuke Nambu, Yoshitaka Tateyama, Shinichi Komaba; writing – review and editing: Yoshihiko Umemoto, Kazuki Ohishi, Daisuke Igarashi, Che-an Lin, Yusuke Nambu, Yoshitaka Tateyama, Shinichi Komaba.Conflicts of interest
We declare that there are no conflicts to declare.Data availability
The experimental data obtained from operando SANS and WANS measurements at the TAIKAN instrument (J-PARC) that support the findings of this study are available from the corresponding authors upon reasonable request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc09600f.
Acknowledgements
We would like to acknowledge Prof. G. Hasegawa and Prof. M. Shibayama for their helpful discussions, which provided useful insights during the preparation of this work. We also would like to acknowledge Mr T. Morikawa for his technical support during the measurements at J-PARC. This work was supported by GteX (grant number: JPMJGX23S4), JSPS KAKENHI (grant numbers: 23H01693, 23K26386, 24H00042, 22H05145, 24K00572, 25K01489), JSPS Core-to-Core Program (grant number: JPJSCCB20240005), JST NEXUS-Ytec (Y2024L0906032), JST FOREST (grant number: JPMJFR202V). The neutron experiment at the Materials and Life Science Experimental Facility of the J-PARC was performed under a user program (Proposal No. 2023C0004 and 2024C0007).Notes and references
- J.-M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 171–179 Search PubMed.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- N. Nitta, F. Wu, J. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- S. Qiu, L. Xiao, M. L. Sushko, K. S. Han, Y. Shao, M. Yan, X. Liang, L. Mai, J. Feng, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2017, 7, 1700403 CrossRef.
- C. Wu, Y. Yang, Y. Zhang, H. Xu, X. He, X. Wu and S. Chou, Chem. Sci., 2024, 15, 6244–6268 RSC.
- B. Chen, Q. Meng, T. Wang, W. Zhang and X. Qiu, J. Power Sources, 2024, 624, 235566 CrossRef CAS.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- R. N. Nasara, W. Ma, S. Tsujimoto, Y. Inoue, Y. Yokoyama, Y. Kondo, K. Miyazaki, Y. Miyahara, T. Fukutsuka, S. kang Lin and T. Abe, Electrochim. Acta, 2021, 379, 138175 CrossRef CAS.
- D. Stevens and J. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- Q. Chen, Q. Wen, C. Li, C. Li, P. Zhao, L. Li, X. Tan, J. Wang, X. Fan, S.-L. Chou and X. Wu, Adv. Mater., 2026, e15495 CrossRef CAS PubMed.
- C. Tang, W. Zhang, Y. Zhang, Q. An, F. Xu, G. Wang and Y. Mai, Adv. Mater., 2025, e11632 Search PubMed.
- A. Kamiyama, K. Kubota, D. Igarashi, Y. Youn, Y. Tateyama, H. Ando, K. Gotoh and S. Komaba, Angew. Chem., Int. Ed., 2021, 60, 5114–5120 CrossRef CAS PubMed.
- D. Igarashi, Y. Tanaka, K. Kubota, R. Tatara, H. Maejima, T. Hosaka and S. Komaba, Adv. Energy Mater., 2023, 13, 2302647 CrossRef CAS.
- D. Igarashi, R. Tatara, R. Fujimoto, T. Hosaka and S. Komaba, Chem. Sci., 2023, 14, 11056–11066 RSC.
- Z. T. Gossage, D. Igarashi, Y. Fujii, M. Kawaguchi, R. Tatara, K. Nakamoto and S. Komaba, Chem. Sci., 2024, 15, 18272–18294 RSC.
- G. Hasegawa, K. Kanamori, N. Kannari, J.-I. Ozaki, K. Nakanishi and T. Abe, ChemElectroChem, 2015, 2, 1917–1920 CrossRef CAS.
- Z. Liu, Z. Lu, S. Guo, Q.-H. Yang and H. Zhou, ACS Cent. Sci., 2023, 9, 1076–1087 CrossRef CAS PubMed.
- R. E. Franklin and J. T. Randall, Proc. R. Soc. A, 1951, 209, 196–218 CAS.
- J. Dahn, W. Xing and Y. Gao, Carbon, 1997, 35, 825–830 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 4428 CrossRef CAS.
- D. Saurel, J. Segalini, M. Jauregui, A. Pendashteh, B. Daffos, P. Simon and M. Casas-Cabanas, Energy Storage Mater., 2019, 21, 162–173 CrossRef.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey, Q. Cai, A. J. Drew and M.-M. Titirici, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- K. Ohishi, D. Igarashi, R. Terata, K. Nakemoto, Y. Kawamura, M. Toshiaki, K. Hiroi, S. Takata and S. Komaba, Accepted in JPS Conf. Proc., 2025 Search PubMed.
- S. Takata, J. Suzuki, T. Shinohara, T. Oku, T. Tominaga, K. Ohishi, H. Iwase, T. Nakatani, Y. Inamura, T. Ito, K. Suzuya, K. Aizawa, M. Arai, T. Otomo and M. Sugiyama, JPS Conf. Proc., 2015, vol. 8, p. 36020 Search PubMed.
- K. Ohishi, D. Igarashi, R. Tatara, I. Umegaki, A. Koda, S. Komaba and J. Sugiyama, ACS Appl. Energy Mater., 2022, 5, 12538–12544 CrossRef CAS.
- K. Ohishi, D. Igarashi, R. Tatara, S. Nishimura, A. Koda, S. Komaba and J. Sugiyama, ACS Phys. Chem. Au, 2022, 2, 98–107 CrossRef CAS PubMed.
- K. Ohishi, D. Igarashi, R. Tatara, Y. Kawamura, K. Hiroi, J. ichi Suzuki, I. Umegaki, S. Nishimura, A. Koda, S. Komaba and J. Sugiyama, J. Phys.: Conf. Ser., 2023, 2462, 012048 CrossRef CAS.
- R. Tatara, D. Igarashi, M. Nakayama, T. Hosaka, K. Ohishi, I. Umegaki, J. G. Nakamura, A. Koda, H. Ohta, R. Palm, M. Månsson, E. J. Kim, K. Kubota, J. Sugiyama and S. Komaba, Chem. Sci., 2025, 16, 19990–20001 RSC.
- Y. Inamura, T. Nakatani, J. Suzuki and T. Otomo, J. Phys. Soc. Jpn., 2013, 82, SA031 CrossRef.
- J. Suzuki, T. Nakatani, T. Ohhara, Y. Inamura, M. Yonemura, T. Morishima, T. Aoyagi, A. Manabe and T. Otomo, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 600, 123–125 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- R. Peverati, Y. Zhao and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 1991–1997 CrossRef CAS.
- M. Valiev and J. H. Weare, J. Phys. Chem. A, 1999, 103, 10588–10601 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. Asher, J. Inorg. Nucl. Chem., 1959, 10, 238–249 CrossRef CAS.
- P. Debye and A. M. Bueche, J. Appl. Phys., 1949, 20, 518–525 CrossRef CAS.
- P. Debye, H. R. Anderson and H. Brumberger, J. Appl. Phys., 1957, 28, 679–683 CrossRef CAS.
- M. Teubner and R. Strey, J. Chem. Phys., 1987, 87, 3195–3200 CrossRef CAS.
- K. Schubert, R. Strey, S. R. Kline and E. W. Kaler, J. Chem. Phys., 1994, 101, 5343–5355 CrossRef CAS.
- C. A. Bridges, X.-G. Sun, J. Zhao, M. P. Paranthaman and S. Dai, J. Phys. Chem. C, 2012, 116, 7701–7711 CrossRef CAS.
- E. M. Reynolds, J. Fitzpatrick, M. O. Jones, N. Tapia-Ruiz, H. Y. Playford, S. Hull, I. McClelland, P. J. Baker, S. A. Cussen and G. E. Pérez, J. Mater. Chem. A, 2024, 12, 18469–18475 RSC.
- G. Hasegawa, M. Hattori, K. Nakanishi and K. Hayashi, Energy Storage Mater., 2025, 80, 104437 CrossRef.
- G. Hasegawa, K. Kanamori, N. Kannari, J. ichi Ozaki, K. Nakanishi and T. Abe, J. Power Sources, 2016, 318, 41–48 CrossRef CAS.
- H. Alptekin, H. Au, A. C. Jensen, E. Olsson, M. Goktas, T. F. Headen, P. Adelhelm, Q. Cai, A. J. Drew and M.-M. Titirici, ACS Appl. Energy Mater., 2020, 3, 9918–9927 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |