
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Why is a dicationic digallene so reactive towards activation of strong covalent bonds? Scope and mechanistic investigations
Antoine
Barthélemy
 ,
Nico Gino
Kub
,
Celine
Regnat
,
Harald
Scherer
and
Ingo
Krossing
*
,
Nico Gino
Kub
,
Celine
Regnat
,
Harald
Scherer
and
Ingo
Krossing
*
Institut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
First published on 2nd February 2026
Abstract
Herein, we investigate the reactivity of the trans-bent Ga⇄Ga double bond in the dicationic digallene [{Ga(dcpe)}2]2+ (dcpe = bis(dicyclohexylphosphino)ethane) as its [pf]− salt ([pf]− = [Al(ORF)4]−; RF = C(CF3)3), which is formed in situ within seconds. Unusually, this digallene is highly reactive towards covalent bonds and oxidatively adds even to strong E–Y σ-bonds, e.g., H–O, H–N, H–C and C–F bonds, under mild conditions, often at room temperature. Their bond activation at any cationic subvalent group 13 compound is unprecedented and the C–H bond activation is the first oxidative addition reported between any subvalent gallium compound and a neutral substrate. The scope and mechanism of the bond activation reactions were experimentally investigated by interaction with selected substrates and via isotope labelling experiments, as well as using high-level quantum chemical calculations. Mechanistically, the pronounced reactivity of the digallene can be attributed to an easily accessible asymmetric conformer with one (Lewis-acidic) planarized and one (Lewis-basic) pyramidalized reactive Ga-site, allowing for cooperative E–Y bond cleavage. In addition, the [2 + 2] cycloaddition of the Ga⇄Ga bond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double and triple bonds was studied: it follows a stepwise, non-concerted reaction mechanism, which allows for the catalytic isomerization of cis-olefins and may serve as the basis for follow-up functionalization reactions.
C double and triple bonds was studied: it follows a stepwise, non-concerted reaction mechanism, which allows for the catalytic isomerization of cis-olefins and may serve as the basis for follow-up functionalization reactions.
Introduction
Although transition metal catalysts are widely employed in industry, their scarcity, high cost, and the environmental burdens associated with their mining continuously drive research towards more sustainable metal alternatives. In this context, low valent main group metal compounds have demonstrated their potential as viable alternatives to transition metal catalysts, as main group elements tend to be more abundant and cost efficient.1–4 With orbital hybridization between valence orbitals getting less pronounced as one descends in any given p-block group, most heavier low valent main group carbene analogues occupy a singlet ground state. In this state, the combination of a filled ns-type orbital and vacant np-type orbital leads to both, electrophilic and nucleophilic properties, which enable insertion into various σ-bonds by oxidative addition.3,4 Analogous to carbenes, heavier low valent group 13 and 14 compounds can dimerize into ditrielenes or ditetrylenes, respectively.5 Yet, the nature of the thereby formed formal double bond differs greatly from typical CC double bonds. Their bonding situation is best described as a double-dative bond with a trans-bent geometry, in which the heavier singlet trielene/tetrylene fragments reciprocally donate electron density from the filled ns-orbital to the empty np-orbital of the adjacent fragment,5–7 as visualized in Scheme 1a. Oligomerization is only suppressed with suitable sterically demanding substituents R, as first demonstrated by Lappert and West with a stable distannene8 and a disilene.9 Considerable follow-up research efforts led to the isolation of a multitude of trans-bent digermene,10 distannene11 and diplumbene structures.8,12 Notably, these heavier ditetrylenes, in particular disilenes and digermenes, readily activate covalent σ- and π-bonds.13,14 After the isolation of the first molecular digallene by Power et al.,15 the chemistry of (neutral) ditrielenes also evolved into a major research field.16–18 However, reports of σ-bond activation reactions with group 13 dimers are still rather scarce.17,19,20 Instead, oxidative addition reactions with subvalent group 13 compounds are usually performed at monomeric complexes, or, if dinuclear, those without a direct group 13 metal–metal interaction. Such compounds are almost invariably neutral or anionic, exhibit high reactivity towards covalent bonds, but require the use of sophisticated ligands with multi-step syntheses.21–26 Selected examples are shown in Scheme 1b, with the anionic aluminyl anions, introduced by the Aldridge-group, ranking amongst the most reactive compounds that even activate C–H and C–C bonds, which is attributed to a high-lying HOMO and a small HOMO–LUMO gap.27
 | ||
| Scheme 1 (a) Orbital interactions of two singlet fragments in a trans-bent double bond for ditetrylenes and ditrielenes.7,28,29 (b) Selected examples for subvalent group 13 complexes used for bond activation reactions (Dipp = 2,6-iPr2C6H3; Ad = adamantyl; 12-c-4 = 12-crown-4).23–26 (c) Scope of this work. The activation of both π-bonds and very strong σ-bonds was investigated with the dicationic digallene [{Ga(dcpe)}2]2+. Published average bond dissociation energies (BDEs) are also included,30–32 which are heavily influenced by the substituents Rn; only rough average values are given. At the bottom, the crystallographically found symmetric and an accessible asymmetric conformer relevant for the cooperative reactivity of the digallene 12+ are shown. Trends of their partial charges and relevant orbital fragments are drawn in for the Ga atoms, as well as the suggested transition state important for the σ-bond activation reactions. | ||
Yet, to allow the progress from mere bond activation/oxidative addition to true catalysis with relevant bonds, the reverse process – the reductive elimination – also needs to be energetically feasible. And, while anionic and neutral subvalent main group complexes typically undergo irreversible oxidative addition reactions impeding catalytic applications with such substrates, a few reductive elimination reactions at cationic gallium complexes have been reported.33–36 However, oxidative additions at cations are generally less favorable due to reduced orbital energies. Thus, a major goal in cationic group 13 chemistry is to identify metal/ligand combinations that enable reversible substrate activation, in order to realize true main group catalysis.37,38 However, bond activation at group 13 cations is still in its infancy, a fact to be overcome with this contribution.29,35,39,40
Favorably, and in contrast to other compounds with trans-bent double bonds requiring time-consuming multi-step syntheses, we recently obtained [{Ga(dcpe)}2][pf]2 ([pf]− = [Al(ORF)4]−; RF = C(CF3)3; dcpe = (bis(dicyclohexyl-phosphino)ethane)) in situ within a matter of minutes by mixing [Ga(PhF)2][pf],41 a stable source of univalent gallium, with the commercially available bisphosphine dcpe in the weakly coordinating solvent oDFB (ortho-difluorobenzene).29 The large, weakly coordinating [pf]− anion33,42 prevents disproportionation of the dicationic trans-bent digallene [{Ga(dcpe)}2]2+ (12+), which is isoelectronic and isostructural to neutral ditetrylenes (Scheme 1a). Note that changing the ligand from dcpe to sterically leaner systems led to the formation of polycationic gallium clusters with Ga–Ga single bonds.37,43 By contrast, the use of increasingly bulkier bisphosphine ligands allows for the formation of either dimeric or monomeric (di)gallenes, which, for example, with dipf as a ligand (dipf = 1,1′-(iPr2P)2Fc), exhibit a temperature-dependent monomer–dimer equilibrium in solution.40 With the latter ligand, monomers and dimers already displayed Si–H and B–H bond activation potential, but the reactivity of the monomer and dimer is hard to separate for mechanistic studies.40
Hence, in this work we targeted reactions with the [{Ga(dcpe)}2]2+ (12+) system that is a stable dimer under all conditions tested (Scheme 1c).† We tested its limit for bond activation towards π and very strong σ bonds. In particular, the presence of an asymmetric conformer with separate donor and acceptor sites, as shown in Scheme 1c, might play a role in the observed bond activation reactions. In favor of this, preliminary work showed that 12+ undergoes [2 + 2] cycloaddition reactions with unsaturated substrates and oxidative addition reactions, albeit only with rather weak S–S and P–H bonds, mimicking to some extent the reactivity of the well-known neutral ditetrylenes.1,29 Its full scope is analyzed here.
Results and discussion
First, we describe all experiments, before analyzing the underlying mechanistic aspects in the second part, further supported by double-hybrid DFT calculations and bonding analyses. However, where sensible, the energetics underlying a given reaction is already included with its first description.Part I – bond activation reactions with the dicationic digallene
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C double and C
C double and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C triple bonds.
To analyze the reaction mechanism of the [2 + 2] cycloaddition reaction, digallene 12+ was reacted with selected alkenes and alkynes to determine whether the reaction follows a stepwise or a concerted mechanism. Trans-deutero-styrene, cis-β-methyl-styrene, cis-3-hexene and ethinyl-cyclopropane were chosen as suitable substrates. The reaction outcomes are summarized in Scheme 2. Unless otherwise stated, the reactions were carried out at room temperature (rt) in oDFB.
C triple bonds.
To analyze the reaction mechanism of the [2 + 2] cycloaddition reaction, digallene 12+ was reacted with selected alkenes and alkynes to determine whether the reaction follows a stepwise or a concerted mechanism. Trans-deutero-styrene, cis-β-methyl-styrene, cis-3-hexene and ethinyl-cyclopropane were chosen as suitable substrates. The reaction outcomes are summarized in Scheme 2. Unless otherwise stated, the reactions were carried out at room temperature (rt) in oDFB.
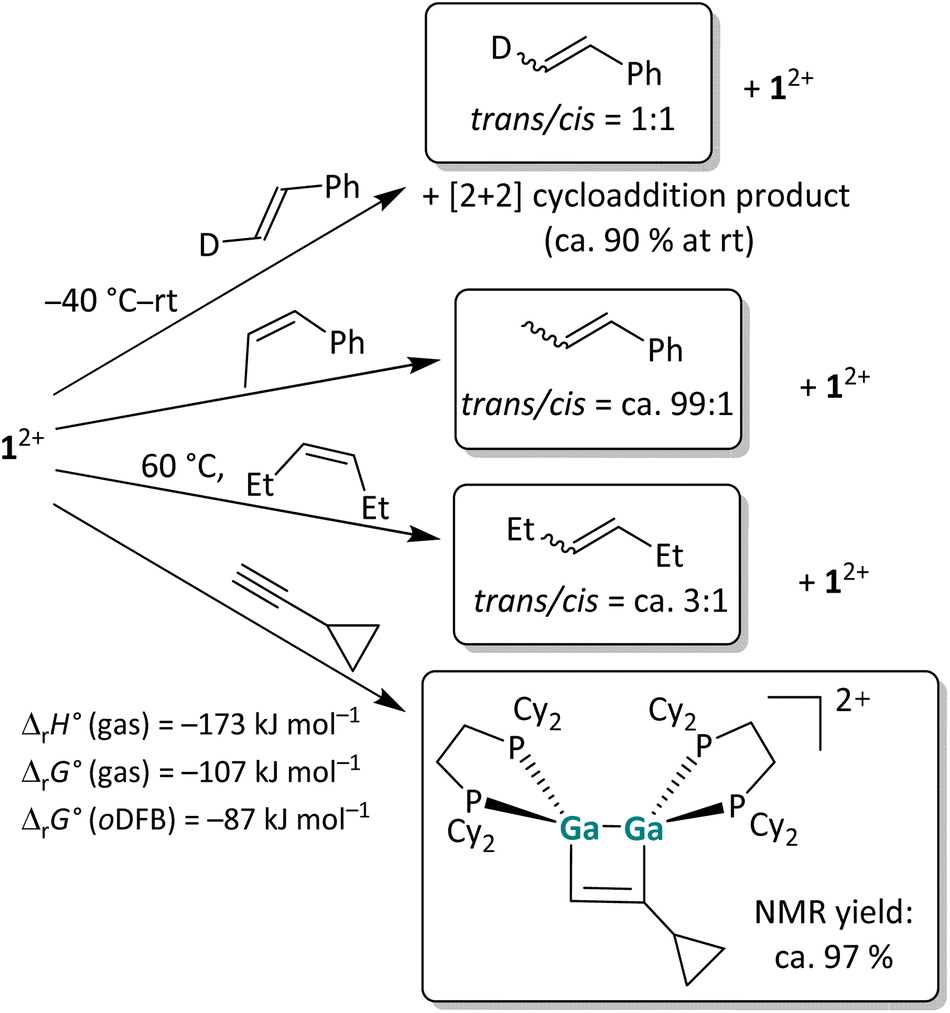 | ||
| Scheme 2 Overview of the reactivity of 12+ towards three olefins with increasing steric protection of the double bond and the “radical clock” ethinylcyclopropane in oDFB. The [pf]− anion is omitted for clarity. The reactions were carried out at rt in oDFB and examined via NMR spectroscopy. The calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB are also given for the cycloaddition reaction (accurate44 DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculations on RI-BP86(D3BJ)/def2-TZVPP optimized structures; εr = 13.38).32 | ||
Trans-deutero-styrene. In the reaction of a dipf-substituted dicationic digallene with styrene, an equilibrium between the starting material and the [2 + 2] cycloaddition product was observed.40 Also with 12+, no full conversion of the olefin is observed and a 9
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the cycloaddition product and the free olefin is obtained at rt in solution. More importantly, 1H NMR spectroscopy reveals that already at −40 °C, before any cycloaddition products are observable, trans-deutero-styrene is converted to a 1:1 mixture of the trans and cis isomers in the presence of 12+ (Section 4.1.1 in the SI). The fast isomerization, even with a sterically unhindered olefin, presents evidence that the [2 + 2] cycloaddition reaction of dicationic digallenes does not follow a concerted but rather a stepwise mechanism. Consequently, the reaction needs to proceed via intermediates where free rotation around the former CC double bond is possible.
:1 ratio between the cycloaddition product and the free olefin is obtained at rt in solution. More importantly, 1H NMR spectroscopy reveals that already at −40 °C, before any cycloaddition products are observable, trans-deutero-styrene is converted to a 1:1 mixture of the trans and cis isomers in the presence of 12+ (Section 4.1.1 in the SI). The fast isomerization, even with a sterically unhindered olefin, presents evidence that the [2 + 2] cycloaddition reaction of dicationic digallenes does not follow a concerted but rather a stepwise mechanism. Consequently, the reaction needs to proceed via intermediates where free rotation around the former CC double bond is possible.
Other olefins. Also, the cis-methyl-styrene and cis-3-hexene substrates yield a mixture of cis- and trans-isomers in the presence of 12+, while no cycloaddition product is observed with these substrates. Note that the olefin isomerization can also be performed with catalytic amounts, i.e. 10 mol%, of 12+, which makes this reaction an example of Lewis-acid catalyzed cis/trans olefin isomerization.45 The observed cis/trans ratios depend on the relative stabilities of the stereoisomers in comparison to one another and imply thermodynamic control. Hence, cis- and trans-deutero-styrene are similarly stable for steric and electronic reasons, while the trans isomers of 3-hexene and especially of β-methyl-styrene are more stable than the respective cis isomers. As a consequence, the cis/trans equilibrium ratio increases in this order as determined by 1H NMR spectroscopy (Scheme 2). Intriguingly, no [2 + 2] cycloaddition products form with these substrates, probably due to steric constraints and the steric bulk of the cyclohexyl groups. Finally, the clean [2 + 2] cycloaddition reaction with the “radical clock” substrate ethinyl-cyclopropane indicates that the cycloaddition does not proceed via radical intermediates, since otherwise a very fast ring opening of the 3-membered ring would be expected.46
Reaction with a butadiene. In addition, dicationic digallenes not only undergo [2 + 2] cycloaddition reactions, but also [2 + 4] cycloaddition reactions, i.e. hetero Diels–Alder reactions,47 as described in Scheme 3.48 The reaction with 2,3-dimethylbutadiene yields a stable digalla-cyclohexene, which is highly exothermic and exergonic as suggested by quantum chemical calculations and notably thermodynamically more favorable than the [2 + 2] cycloaddition with the non-conjugated olefin 1-hexene (for comparison: ΔrH° (gas) = −107 kJ mol−1, ΔrG° (gas) = −41 kJ mol−1, ΔrG° (oDFB) = −31 kJ mol−1).29
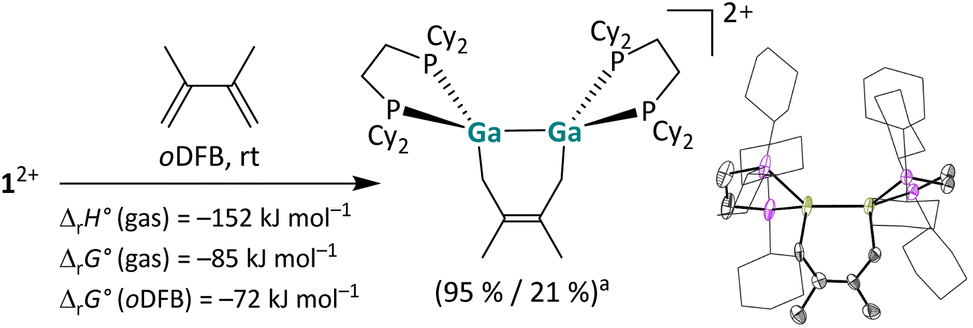 | ||
| Scheme 3 [2 + 4] Cycloaddition of 12+ with 2,3-dimethylbutadiene and the molecular structure of the product [{Ga(dcpe)}2(C6H10)]2+ in the solid state (a NMR yield/isolated yield). The [pf]− anions and hydrogen atoms are omitted for clarity. The cyclohexyl groups are depicted in wireframe for simplicity. Thermal ellipsoids are set at the 50% probability level. The calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB are also given (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures; εr = 13.38).32 | ||
We already reported a [1 + 4] cycloaddition reaction of a 1,3-diene with a monocationic, gallene monomer.35 Furthermore, [2 + 4] cycloaddition reactions are well known for neutral digallenes,49 thus emphasizing the structural and electronic relationship of [{Ga(dcpe)}2]2+ between these substance classes.
 | ||
| Scheme 4 Overview of the reactivity of 12+ towards ethanol, phenol, n-butylamine, aniline, and diphenylamine in oDFB. Unless otherwise stated, the reactions were carried out at rt (a NMR yield/isolated yield). The molecular structures of [H{Ga(dcpe)}2OEt]2+, [H{Ga(dcpe)}2OPh]2+, [H{Ga(dcpe)}2NHPh]2+ and [H{Ga(dcpe)}2NPh2]2+ are included. The [pf]− anions and hydrogen atoms – except those bound to Ga – are omitted for clarity. The cyclohexyl groups are depicted in wireframe for simplicity. Thermal ellipsoids are set at the 50% probability level. The calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB are also given (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures, εr = 13.38).32 | ||
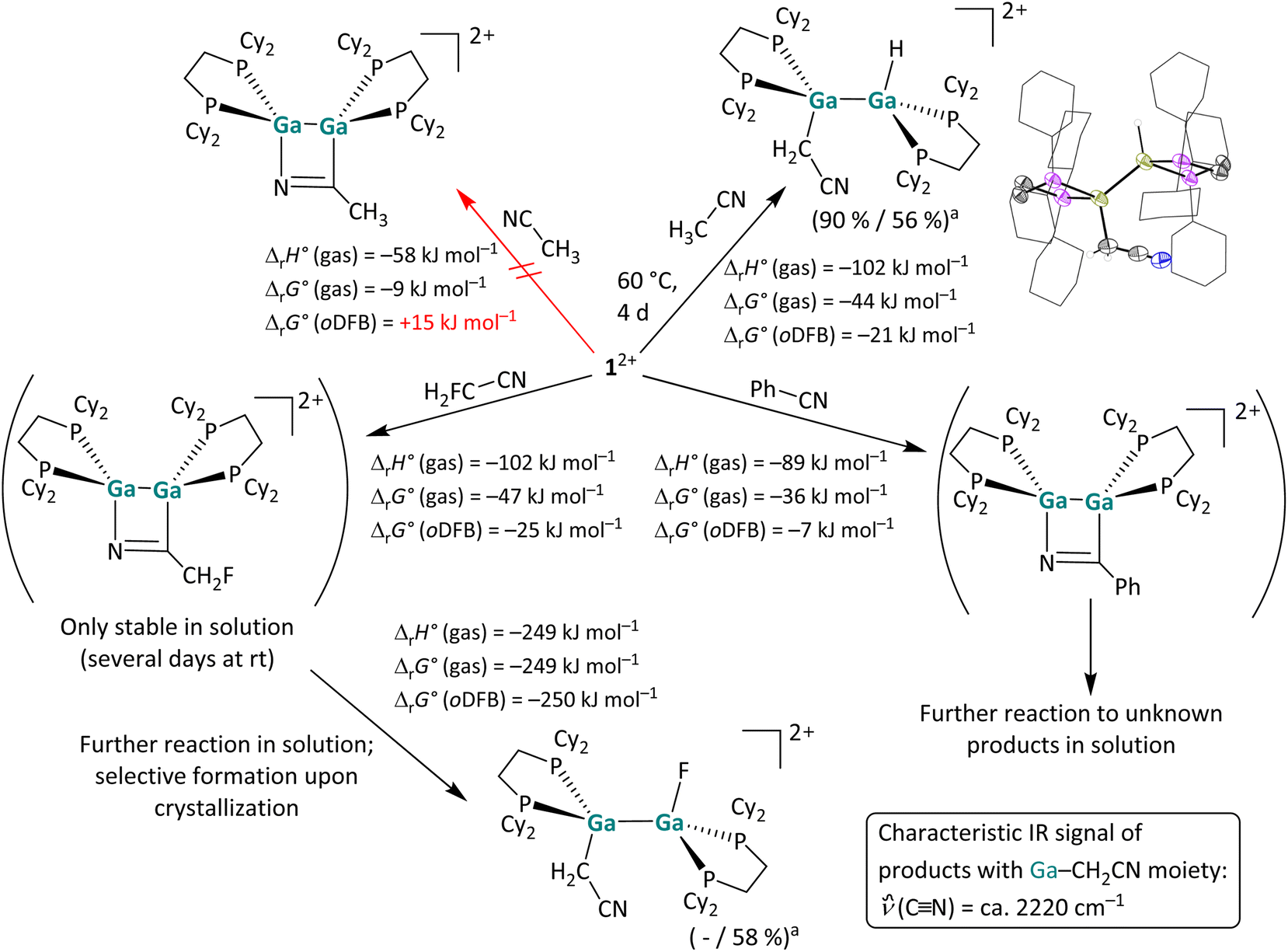 | ||
| Scheme 5 Overview of the reactivity of 12+ towards FH2C–CN, Ph–CN and H3C–CN. Unless otherwise stated, the reactions were carried out at rt (a NMR yield/isolated yield) in oDFB. The hypothetical [2 + 2] cycloaddition product with acetonitrile is also shown as well as the actually formed C–H bond activation product. The molecular structure of the resulting [H{Ga(dcpe)}2CH2CN]2+ is included. The [pf]− anions and hydrogen atoms – except those bound to Ga and the adjacent methylene group – are omitted for clarity. The cyclohexyl groups are depicted in wireframe for simplicity. Thermal ellipsoids are set at the 50% probability level. The calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB are given (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures). | ||
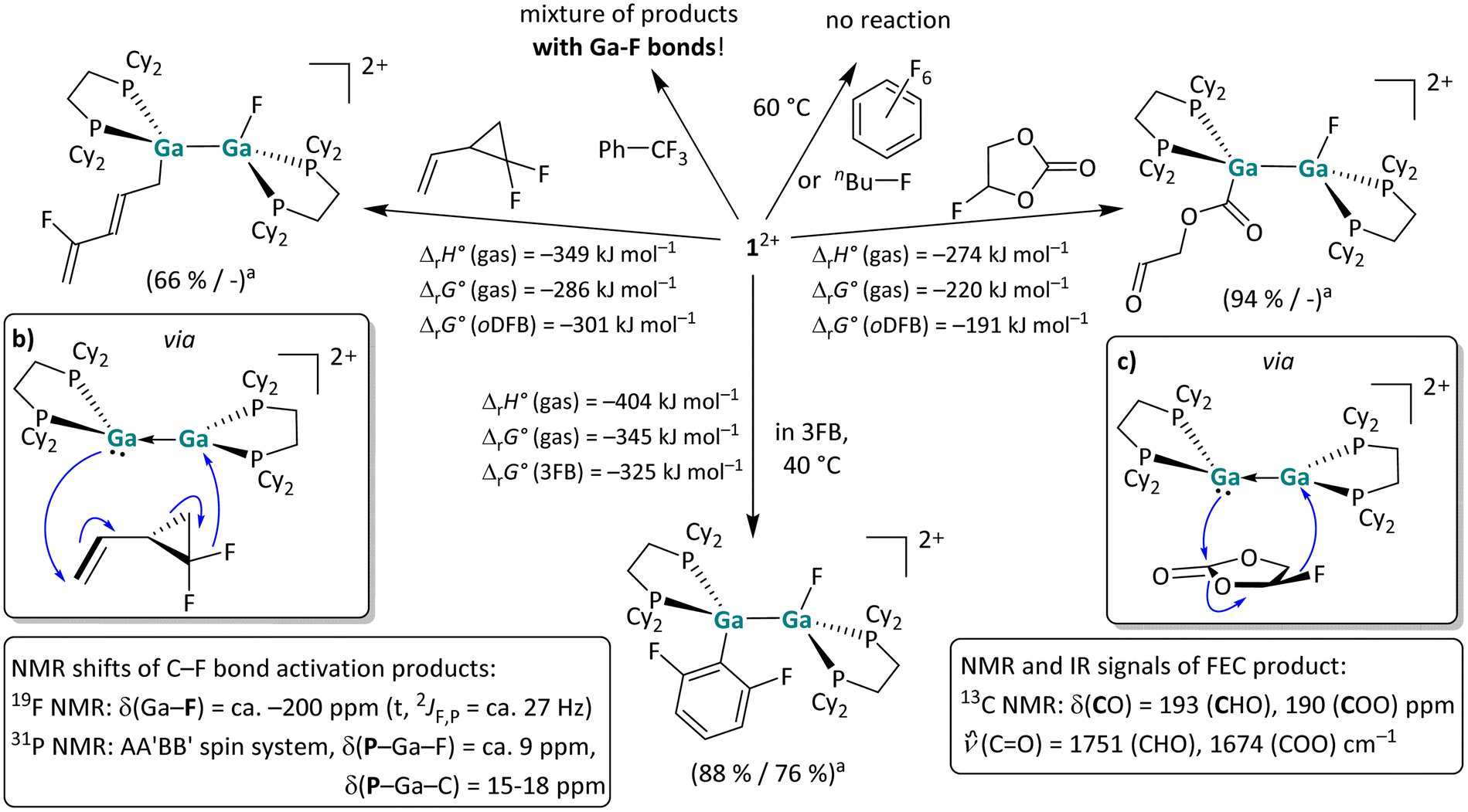 | ||
| Scheme 6 (a) Overview of the reactivity of 12+ towards hexafluorobenzene, 1-fluorobutane, trifluorotoluene, 1,1-difluoro-2-vinylcyclopropane and fluoroethylenecarbonate (FEC) in oDFB and towards 3FB in the neat solvent (a NMR yield/isolated yield). The [pf]− anion is omitted for clarity. Unless otherwise stated, the reactions were carried out at rt. It should be noted that the C–F bond activation with 1,1-difluoro-2-vinylcyclopropane is somewhat unselective and that the structure of the main product is shown. The suggested electron flow in the adducts of 12+ and 1,1-difluoro-2-vinylcyclopropane (b), as well as of 12+ and FEC (c) accounts for the formation of the respective products. | ||
H–E (E = N, O) bond activation reactions. The digallene 12+ reacts both with aliphatic and aromatic alcohols and amines, as summarized in Scheme 4. To the best of our knowledge, the H–O bond activation reactions lead to the first examples of group 13 dimers with the structural motif H–E–E–Oalkoxy/aryloxy (E = group 13 metal). Interestingly, the simple reduction of the polar amine/alcohol by the subvalent digallene and with evolution of dihydrogen according to RnE–H + GaI → RnE–GaIII + 0.5 H2 (E = N, O), was never observed. By contrast, an oxidative addition reaction of the digallene into the H–O bond of ethanol and phenol takes place already at rt. The insertion of 12+ into the H–N bond also occurs at rt for n-butylamine and aniline, but the addition of diphenylamine required additional heating of the reaction mixture to 40 °C for 16 hours. Cleavage reactions of O–H bonds have previously been reported with neutral, monomeric NHC analogues of Al(I)50 and Ga(I).36 However, the addition of a H–O bond across a ditrielene double bond is, to the best of our knowledge, unprecedented. Similarly, the activation of both primary and secondary amines has been reported via neutral ditetrylenes,14,51 and the activation of ammonia has been demonstrated with anionic Al(I) compounds and a neutral digallene.19,52 Yet, the activation of H–N bonds with ditrielenes is rather rare.17,19
Characterization. 1H NMR and IR spectroscopy are useful tools to monitor the E–H bond activation: the H–Ga proton resonates at ca. 4.6 ppm and exhibits a triplet splitting pattern with 2JH,P ≈ 20 Hz, while the H–Ga bond produces a broad band at ca. 1850 cm−1 in vibrational spectra. The scXRD measurements reveal an anti-periplanar geometry of the products, with H–Ga–Ga–E torsion angles ranging from 114° to ca. 175°. It can be assumed that the large range of torsion angles can be attributed to different dispersion interactions between the Cy rings and the respective organic substituents of the substrate. Furthermore, the trigonal planar coordination of the N atom in the activation products of H2NPh and HNPh2 is noteworthy and agrees with a formal sp2 hybridization of the N atoms in the products.
All reactions in Scheme 4 are exothermic and exergonic, with O–H insertions including the more electronegative atom being more favored than N–H activations. It should be pointed out that the HNPh2 and H2NnBu activations are favored in solution only by −44 to –48 kJ mol−1, which might hint at some reversibility.
Reactions with (un)functionalized nitriles. Next, the reactivity of 12+ towards a C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond was investigated by reactions with acetonitrile, fluoroacetonitrile and benzonitrile (Scheme 5). Of special interest was whether one would observe simple coordination compounds with the nitrile donor, like in the [Ga(NCMe)2]+ complex salt recently reported,53 or whether one would observe a cycloaddition or even another bond activation reaction. The NMR spectra indicate that benzonitrile and 12+ form a [2 + 2] cycloaddition product at rt in a slightly exergonic reaction, which decays to unknown decomposition products within 24 hours, while no reaction was observed between 12+ and acetonitrile at rt. Interestingly, after increasing the reaction temperature to 60 °C the C–H insertion product rather than the coordination or [2 + 2] cycloaddition product with the CN triple bond was observed. DFT calculations suggest that the formation of a [2 + 2] cycloaddition product is thermodynamically unfavored (ΔrG° (oDFB) = +15 kJ mol−1), as is the simple formation of a coordination compound (ΔrG° (oDFB) = +14 kJ mol−1; see Fig. 4).
N triple bond was investigated by reactions with acetonitrile, fluoroacetonitrile and benzonitrile (Scheme 5). Of special interest was whether one would observe simple coordination compounds with the nitrile donor, like in the [Ga(NCMe)2]+ complex salt recently reported,53 or whether one would observe a cycloaddition or even another bond activation reaction. The NMR spectra indicate that benzonitrile and 12+ form a [2 + 2] cycloaddition product at rt in a slightly exergonic reaction, which decays to unknown decomposition products within 24 hours, while no reaction was observed between 12+ and acetonitrile at rt. Interestingly, after increasing the reaction temperature to 60 °C the C–H insertion product rather than the coordination or [2 + 2] cycloaddition product with the CN triple bond was observed. DFT calculations suggest that the formation of a [2 + 2] cycloaddition product is thermodynamically unfavored (ΔrG° (oDFB) = +15 kJ mol−1), as is the simple formation of a coordination compound (ΔrG° (oDFB) = +14 kJ mol−1; see Fig. 4).
The observed activation of a C–H bond, representing the initial step of C–H functionalization reactions for the introduction of functional groups to abundant, simple starting materials, is even more challenging and has long been the domain of transition metal chemistry.54 Considerable progress in the field of subvalent main group element chemistry revealed that certain subvalent main group complexes are also able to cleave C–H bonds, e.g. neutral or anionic Al(I) compounds.25,50,55 However, the reaction with MeCN described herein is, to the best of our knowledge, the first example of an oxidative addition of GaI into a C–H bond of a neutral substrate. Such a bond activation was only described between an anionic GaI-NHC analogue and an imidazolium cation.56
Intriguingly, the reaction with monofluoro-acetonitrile does not initially yield the analogous C–H or C–F bond activation product, but instead the [2 + 2] cycloaddition product formed in a very fast reaction at rt (Scheme 5), according to NMR spectroscopy: the product has two pairs of chemically non-equivalent P atoms, the CH2F group is still intact, and the 13C signal of the adjacent carbon atom is significantly low-field shifted. Notably, the 13C shift is comparable to the shift of the formally sp2-hybridized carbon atom in the [2 + 2] cycloaddition product of 12+ with cyclooctyne (ca. 215 ppm vs. 180 ppm).29 Unfortunately, crystallization of the [2 + 2] cycloaddition product with H2FC–CN did not yield suitable crystals for scXRD. However, NMR spectra of the dissolved crystalline precipitate revealed that the product reacts further and yields the C–F activation product (Scheme 5).‡ Concomitantly, we found that the [2 + 2] cycloaddition product also slowly decomposes in solution at rt, forming a range of unidentified compounds, including the simple C–F activation product. It can be concluded that, with H2FC–CN, cycloaddition and C–F bond activation are competing reaction pathways. DFT calculations reveal that the C–F bond cleavage is remarkably exergonic (Scheme 5), leading to the conclusion that the cycloadduct is the kinetic product, while the C–F bond insertion product can be considered the thermodynamic product. It should be noted that the [2 + 2] cycloaddition products of PhCN and H2FCN with 12+ are considerably less stable than the cycloaddition products of 12+ with alkenes and alkynes, which is reflected by the less exergonic or even endergonic formation of the nitrile cycloadducts (cf. the values in Schemes 2, 3, 5 and ref. 29). Nevertheless, the observed C–F bond activation with CH2FCN prompted us to explore the reactivity of 12+ towards C–F bonds in nitrile-free substrates.
C–F bond activation reactions. Fluorinated organic compounds are interesting and challenging compounds for bond activation reactions due to the inherent strength of C–F bonds.57 Recently, C–F bond cleavage in fluorinated benzenes with a neutral, dinuclear GaI compound as well as neutral dialumenes was reported.18,22 As summarized in Scheme 6, the digallene 12+ was reacted with several fluorinated substrates. No reaction was observed with hexafluorobenzene and 1-fluorobutane, whereas the reaction with trifluorotoluene yielded a mixture of Ga–F-containing products. Gratifyingly, the somewhat surprising molecular structures of C–F bond activation products with 1,1-difluoro-2-vinylcyclopropane and fluoroethylenecarbonate (FEC) could be determined unambiguously via NMR spectroscopy and are both shown in Scheme 6. In addition, 12+ readily and selectively activates the central C–F bond in 1,2,3-trifluorobenzene (3FB) under mild conditions at room temperature. For all the substrates investigated herein, we found that characteristic triplets (2JF,P ≈ 27 Hz) in the 19F NMR spectra at ca. −200 ppm serve as an indicator of a successful fluoride abstraction. The respective 31P{1H} spectra are characterized by a higher order AA′BB′ spin system with chemical shifts at 15–18 ppm and at ca. 9 ppm for the P–GaII–C and P–GaII–F phosphorus atoms respectively.
As expected, the 13C NMR shifts of the Ga-bound carbon atom are less characteristic and depend on the nature of the substituent: for the C–F bond activation products reported in this work, they range from 20 to 114 and 190 ppm for the alkyl, aryl and acyl substituents, respectively (Sections 4.12–4.15 in the SI). The results presented in Scheme 6 strongly suggest that the substrate coordination towards the dicationic digallene 12+ is a prerequisite for C–F bond cleavage. While hexafluorobenzene and 1-fluorobutane are very poor ligands, trifluorotoluene, 1,1-difluoro-2-vinylcyclopropane and FEC can coordinate to metal cations through their comparatively electron rich aromatic π-system, double bond and carbonate group, respectively. It is reasonable to assume that the coordination of the substrate is followed by formal F− transfer, which is only enabled by the close Ga–F proximity. In this context, it is important to mention that the Ga–F bond is the strongest gallium–element single bond (ca. 580 kJ mol−1),30,32 accounting for the pronounced thermodynamic driving force of the F− transfer reactions (see the calculated thermodynamic values in Scheme 6). Consequently, the C–F bond cleavage with trifluorotoluene, leaving a fluorine atom on the first gallium atom and a –CF2Ph moiety on the second gallium atom, is also calculated to be highly exothermic and exergonic (ΔrH° (gas) = −232 kJ mol−1, ΔrG° (gas) = −159 kJ mol−1, ΔrG° (oDFB) = −150 kJ mol−1). The initial product probably undergoes side reactions due to the remaining and reactive Cbenzyl–F bonds, accounting for the observed mixture of products. A clean C–F bond cleavage without accompanying rearrangement reactions was observed for the reaction of 12+ and 3FB after four days at 40 °C (Scheme 6, bottom). The molecular scXRD structure of the product is well reproduced by DFT calculations and is in accordance with NMR spectroscopic results as shown below in Fig. 5, in Part II. Note that [Ga(PhF)2][pf] without a donor ligand is compatible with 3FB.53 In line with previous transition metal mediated reactions, 12+ selectively cleaves the central Caromatic–F bond, confirming that F substituents at ortho positions activate a Caromatic–F bond.58–61 One of the remaining fluorine atoms at the ortho-position is oriented almost vertically above the Ga–Ga bond (Fig. 5b and c), thereby probably stabilizing the formal gallium cations with its free lone pair orbitals (intramolecular CorthoF–Ga distance: ca. 320 pm).
Part II – understanding the reactivity of the dicationic digallene
The reactions with substrates that bear lone pair orbitals at the reactive bond (e.g., N–H and O–H, but also NC–CH3 or F–C), suggested the chance that, prior to the reaction, a substrate/dimer adduct might be relevant. An asymmetric conformer with one more accessible planarized Ga atom may serve as an “anchor” for the substrate, while the other more pyramidal Ga atom could cooperatively perform the subsequent oxidative addition. These hypotheses prompted us to first analyze the frontier MOs (FMOs) of 12+ and scan for other possible conformers, potentially relevant for adduct formation. | ||
| Fig. 1 Comparison of the frontier molecular orbitals (FMOs) of monomeric and dimeric low valent cationic GaI compounds, using the example of the monomeric [Ga(dtbpf)]+ (1,1′-bis(ditertbutylphosphino)ferrocene) and dimeric [{Ga(dcpe)}2]2+ systems calculated at the RI-BP86(BJ)/def2-TZVPP level of theory (isosurface value: 0.05 au). The HOMO and HOMO(−1/−2) of [Ga(dtbpf)]+ are ligand-centered and only have small coefficients on the Ga atom. It should be noted that the HOMO–LUMO gap is even greater for the hypothetical [Ga(dcpe)]+ monomer (HOMO: −8.34 eV; LUMO: −5.61 eV; gap: 2.74 eV). | ||
Apart from the accessible FMOs of 12+, the available conformations of the digallene in solution need to be considered in more detail. Interestingly, the asymmetric conformer shown in Scheme 7 is less stable than the symmetric, crystallographically found conformer only by 10 kJ mol−1. With ΔrG° = –RTlnK, this implies that K = 10−1.7 at 25 °C, or that out of 50 digallenes, one is in the asymmetric conformation. Note that such a briefly mentioned asymmetric conformation for [{Ga(dipf)}2]2+ is considerably higher in energy at +45 kJ mol−1 and, hence, almost irrelevant for reactions.40 The conformational change is well reflected in the bond angles around the Ga atoms: while the sum of the bond angles increases from 310° in the symmetric conformer to 331° for the planarized Ga atom, it decreases to 295° for the pyramidalized Ga atom. The QTAIM (Quantum Theory of Atoms in Molecules) charges indicate the planarized Ga atom to have a slightly higher positive charge than in the symmetric conformer (0.36e vs. 0.34e), while the pyramidal geometry of the second Ga atom indicates the presence of an electron lone pair and thus a more negative partial charge (0.32e). However, the HOMO/LUMO positions and the Ga–Ga bond length are barely affected by this conformational change.
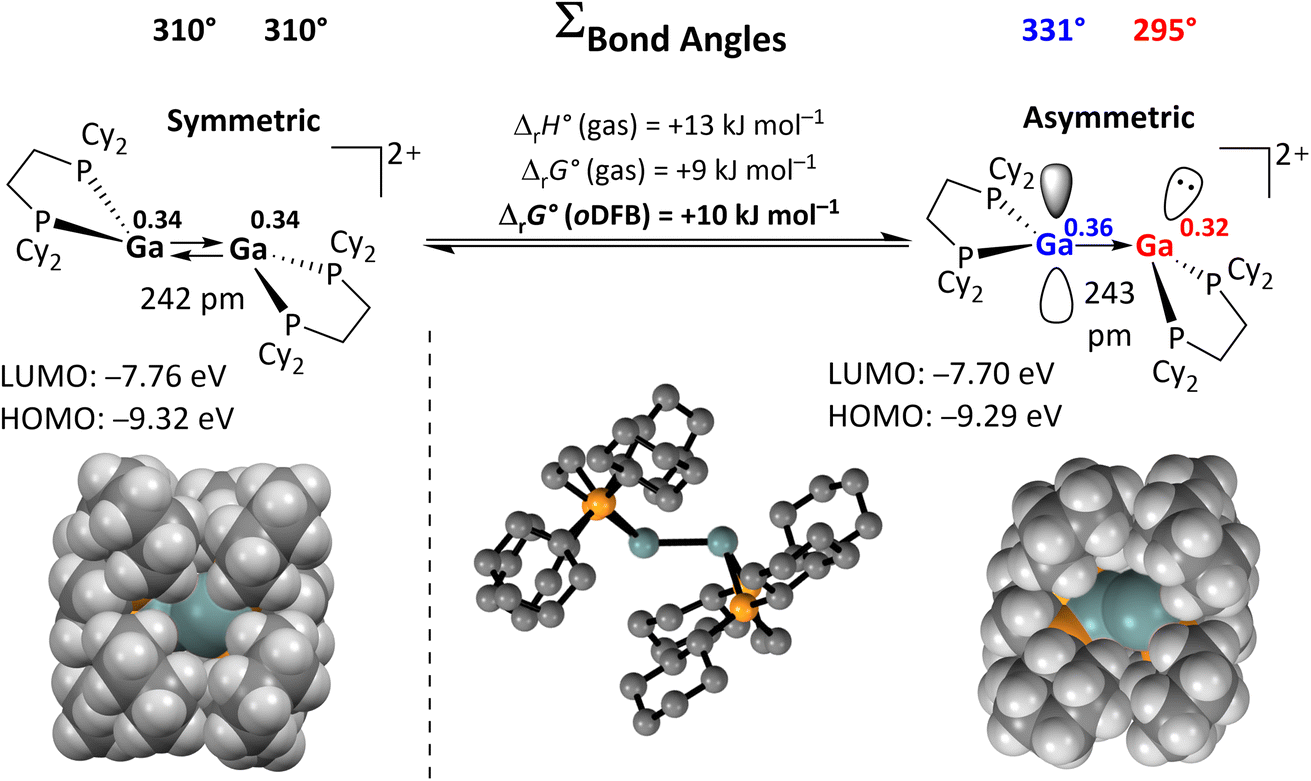 | ||
| Scheme 7 Calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB for the conversion of the symmetric conformation of 12+ into the asymmetric conformation (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures; εr = 13.38).32 The space-filling model and selected calculated parameters for both conformers are also shown, including QTAIM charges for the Ga atoms (superscripts). | ||
The space-filling models of the optimized structures for both conformers nicely illustrate that the Ga atoms in the asymmetric conformer are more accessible (Scheme 7), probably indicating that the conformational change may facilitate the reaction with a given substrate for steric reasons. Hence, although the symmetric global minimum conformer was chosen as a starting point for the calculated thermodynamic values throughout this work, it should be kept in mind that the asymmetric conformer is available and may also serve as the reactive species.
C double bonds to Ga–Ga and C–C single bonds.
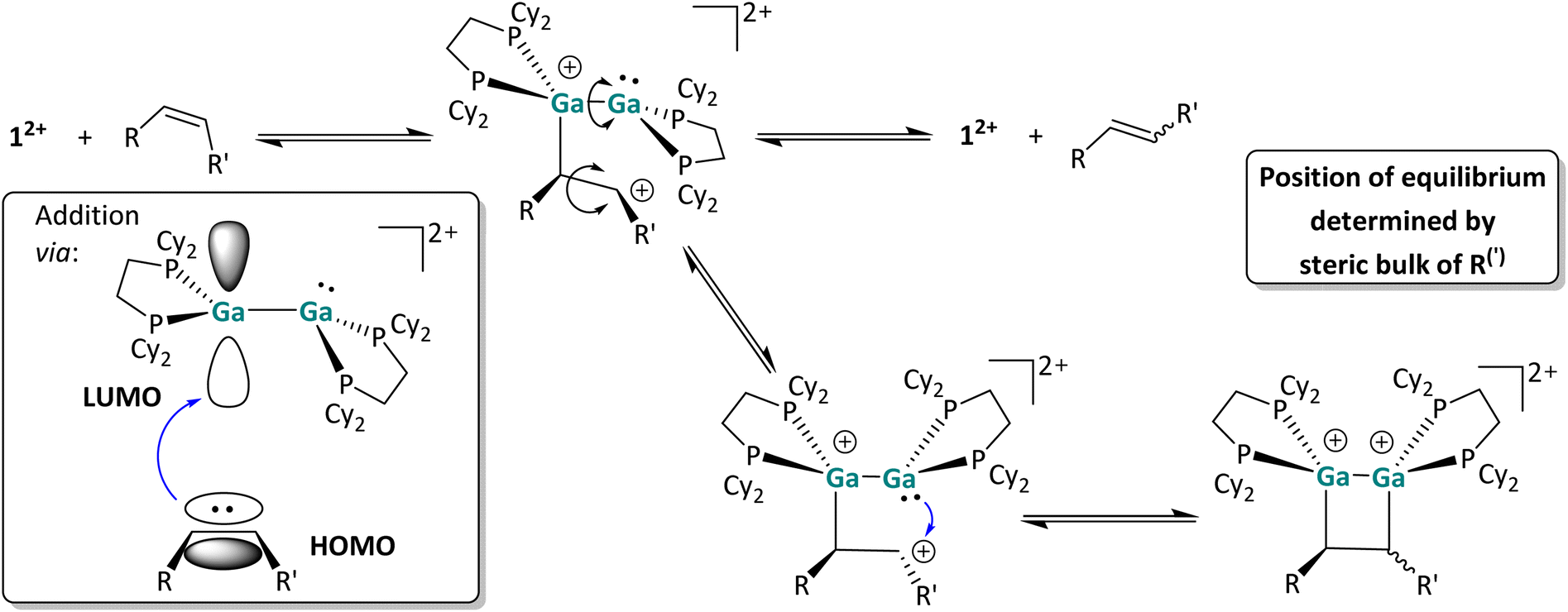 | ||
| Scheme 8 Postulated reaction mechanism of the [2 + 2] cycloaddition reaction of 12+ with CC double bonds. For the sake of clarity, the [pf]− anion is omitted and, for chiral species, only one enantiomer is shown. The most relevant postulated orbital interaction between 12+ and an olefinic substrate is also shown schematically. | ||
In agreement with the low-lying frontier orbitals, the digallene most likely acts as an electrophile in this reaction step, accepting electron density from the olefin and forming a carbocation following Markovnikov's rule. In the resulting carbocation intermediate, rapid rotation around the former CC double bond is possible, accounting for the formation of different stereoisomers in the reaction course. This short-lived carbocation intermediate could not be identified via in situ low temperature NMR spectroscopy; instead, as already described, the cis/trans isomerization was observed immediately in reactions performed at temperatures as low as −40 °C (around the melting point of oDFB), indicating a very low activation barrier for the initial addition of the olefinic species. The carbocationic intermediate next can follow two reaction pathways: it can either undergo ring closure, yielding the [2 + 2] cycloaddition product, or regenerate the dicationic digallene and the (isomerized) olefin via C–Ga bond cleavage. Ring-closure requires rotation around the Ga–Ga bond of the first olefin adduct. Apparently, all species are in equilibrium with one another, and the steric demand of the substrate as well as the capability of the residue to stabilize the carbocation determine whether the [2 + 2] cycloaddition reaction is complete (1-hexene), incomplete (styrene) or does not take place at all (cis 3-hexene and cis methylstyrene).
 | ||
| Fig. 2 (a) Computationally analyzed H–O and H–N bond activation reactions of 12+ with the model substrates HNMe2, H2NMe and HOMe. (b) Energy landscape and intermediates for a hypothetical mechanism of the H–E (E = O, N) bond activation reaction of HOMe (red line), H2NMe (blue line) and HNMe2 (black line) with 12+ (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures; all values are given in kJ mol−1). The Gibbs free energies were calculated for an oDFB solution (εr = 13.38).32 | ||
With the small NHMe, NMe2 and OMe substituents, the initially formed syn-periplanar addition products C are similarly stable or even more stable than the anti-periplanar addition products D. The calculated values are in agreement with the observed reaction rates, as H2NBu and H2NPh react within 30 to 60 minutes, while HOEt and HOPh, react within seconds at rt. However, it should be kept in mind that the exact activation barriers are probably highly influenced by steric effects and dispersion interactions. In line with this, the reaction rate with HNPh2 is considerably slower.
In order to better understand the fundamental electronic structure of the reactive 12+/substrate conformation, it was further analyzed with QTAIM and EDA-NOCV calculations (energy decomposition analysis with the combination of natural orbitals for chemical valence). The small molecules MeOH and Me2NH were chosen as model substrates. The results, summarized in Fig. 3, reveal that the coordination of the substrates leads to significant Ga–Ga bond elongation (15.1 pm and 7.5 pm for the HNMe2 and HOMe complexes, respectively), H–E bond weakening (elongation by 1.5 and 0.2 pm) and considerably more positive charge residing on the coordinating GaI atom, compared to the free, symmetric conformer of 12+ (Δ charge = 0.18 and 0.12 e). Concurrently, the second Ga atom (Δ charge = −0.09 and −0.06) and the donor atom E (Δ charge = −0.07 and −0.06) become more negatively polarized than in the free species. Note that the charge redistribution calculated for the conformer change of the digallene in the absence of a ligand (Scheme 7) is greatly intensified upon substrate coordination. The effects are more pronounced for the Me2NH adduct than with HOMe, agreeing with the generally stronger donor character of amines compared to the analogous alcohols.
 | ||
| Fig. 3 (a) Calculated structures and comparison of selected structural/bond parameters from QTAIM analysis in the Lewis acid–base complexes of 12+ and the model substrates HOMe/HNMe2. Δ = difference in the QTAIM partial charge after coordination of the Lewis base. (b) EDA-NOCV results for the two complexes using 12+ and HOMe or HNMe2 as interacting fragments. The shapes of the deformation densities Δρ(1)–(2) as obtained by the EDA-NOCV analysis, correspond to ΔEorb(1)–(2) for the interaction between 12+ and HNMe2. The fragment orbitals of 12+ and HNMe2 are shown exemplarily (isosurface values: 0.0008 au for Δρ(1)–(2), 0.06 au for the molecular orbitals of 12+, 0.08 au for the HOMO of HNMe2 and 0.05 au for the LUMO(+1) of HNMe2). All other interactions are shown in Section 6.2 of the SI. The direction of the charge flow of the deformation densities is green → yellow. All calculations were performed at the RI-BP86(D3BJ)/def2-TZVPP level of theory; the orbital interaction energies ΔEorb(1)–(2) (for HNMeI2 and HOMeII) are given in kcal mol−1 instead of kJ mol−1 for better comparability with other EDA-NOCV analyses in the literature. (c) Calculated transition state structures of the H–O and H–N bond breakage for HOMe and HNMe2, including selected structural parameters. The value Grel refers to the Gibbs free energies of the transition state structures relative to the free substrates and 12+. | ||
In accordance with the described charge redistribution in Fig. 3, the electrostatic interactions between 12+ and HOMe/HNMe2 make up the strongest contribution to the total attractive interaction, as confirmed by the EDA-NOCV analysis (ca. 50%, Section 6.1 in the SI). By contrast, the dispersion interactions between HOMe/HNMe2 and 12+ are considerably weaker, due to the small organic substituents in the chosen model substrates.
As expected, the electron donation from the HOMO(−1) of the ligand to the LUMO of the asymmetric digallene is the most relevant interaction (Fig. 3b). Interestingly, the second most important interaction is characterized by a reverse electron flow: the HOMO of the asymmetric digallene, which is mainly located on the non-coordinated Ga atom, donates electron density to the LUMO(+1) of the substrates, i.e., the σ*(H–E) orbitals, thus weakening the H–E bonds. Consequently, the cationic digallene primarily acts as a Lewis acid towards substrates with H–E bonds; however, its effect as an electron donor is also relevant. In summary, and as already sketched in Scheme 1b, the EDA-NOCV analysis underlines the importance of both unoccupied and occupied orbitals of 12+ in interactions with σ-bonds and thus confirms the cooperative bond activation mode of the asymmetric conformer of 12+. Notably, also the planarized/pyramidalized asymmetric geometry of the two Ga atoms is retained in the structure of the H–E bond breakage transition states shown in Fig. 3c. This results from the described electron flow.
However, the activation of aromatic substrates may be more complicated than described in Fig. 2. Unlike with the aliphatic substrates, a monomeric, H–Ga containing intermediate was observed via NMR spectroscopy during the reactions of 12+ with diphenylamine. The monomeric species is identified on the basis of a singlet in the 31P{1H} NMR spectrum and its large 2JH,P coupling constant of 50 Hz, which is rather indicative of a GaIII–H species.29,39 By contrast, the 2JH,P coupling constants in the dimeric GaII addition products are significantly smaller (ca. 20 Hz, see Scheme 4). Since the presence of the electron-rich aromatic π-system probably plays a crucial role, we propose the initial formation of an encounter complex, as visualized in Scheme 9a. The affinity of 12+ towards aromatic π-systems is further discussed in the context of the reaction with 3FB (intermediate B in Fig. 5).
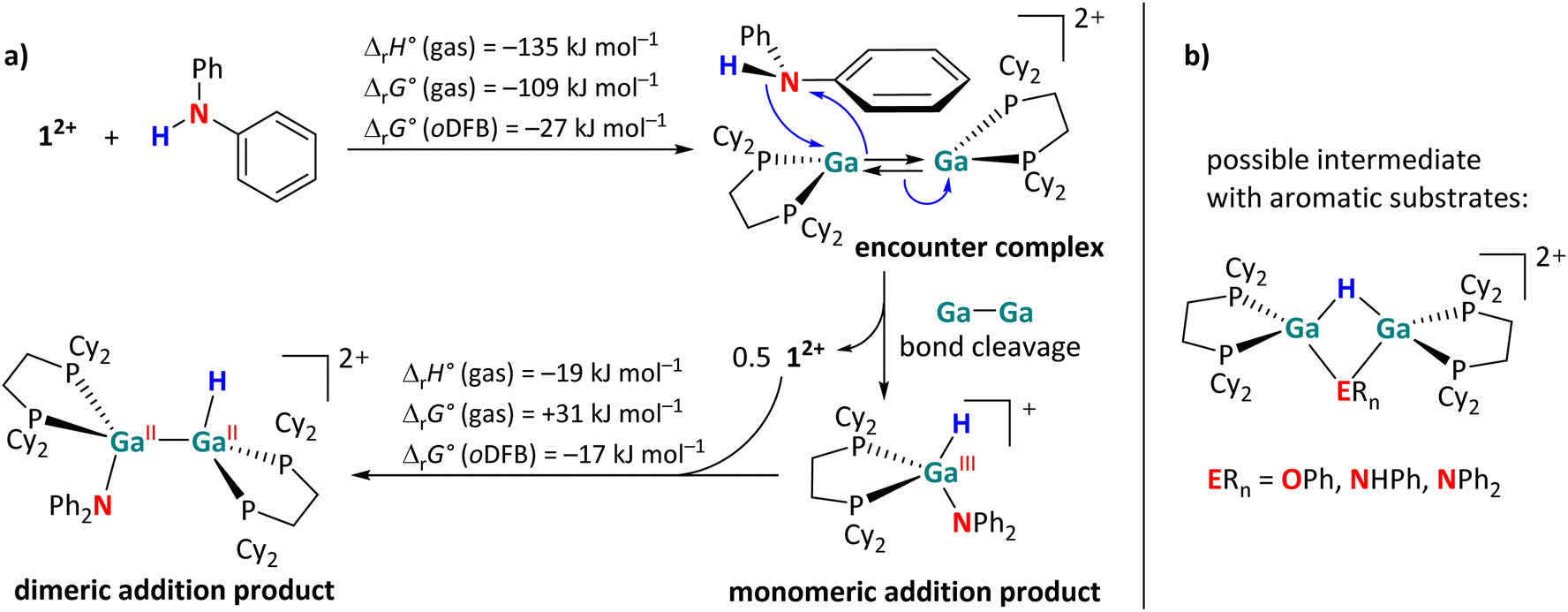 | ||
| Scheme 9 (a) Sketch of the proposed, main activation mechanism of HNPh2, with calculated reaction enthalpies and Gibbs free energies in the gas phase and in oDFB (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures). (b) Possible structure of NMR-spectroscopically observed intermediates during the E–H (E = O, N) bond activation reactions with the aromatic substrates HOPh, H2NPh and HNPh2. Taking into account that the 31P NMR and the 1H NMR shifts of the P–Ga–H units in these intermediates are very similar, it cannot be ruled out that the ERn groups act as bridging ligands with their aromatic π-systems or that the ERn groups are only weakly associated. | ||
Due to the close proximity of the N–H bond to only one Ga atom in the encounter complex, the –NPh2 and –H fragments are transferred to the same Ga atom, while the other Ga+ cation interacts with the π-system of the aromatic substituent and reforms 12+ after dissociation (Scheme 9a). The monomeric addition product further reacts with 12+, ultimately leading to the dimeric addition products shown in Scheme 4. Interestingly, the formation of a monomeric intermediate was not observed during the H–P activation of HPPh2.29 This is most likely due to the fact that phosphines are (towards Ga+) stronger σ-donors than amines, making the η6 coordination mode very uncommon for arylphosphines.62
In addition, another intermediate can be identified during the reactions of 12+ with HOPh, H2NPh and, to a smaller extent, with HNPh2. The singlets in the 31P{1H} NMR spectrum, display a JH,P coupling constant of ca. 27 Hz. Since these species are directly converted to the final addition product, when no more 12+ is present in solution, we suggest these intermediates to have a symmetric, but dimeric structure as proposed in Scheme 9b. The formation mechanism of these bridged species is not entirely clear. They could form after a H atom migration of the initially formed syn-periplanar product, which would allow the formation of the anti-periplanar product without the rotation around the sterically congested Ga–Ga bond.
:1 mixture of acetonitrile and acetonitrile-d3. As revealed by 1H, 13C HMBC NMR spectroscopy, two different products with a Ga–H bond and an adjacent Ga–C bond were formed. Since the other 1H, 13C and 31P chemical shifts are very similar or even virtually identical in both compounds, this can only be explained by the formation of mixed species, i.e., products with H–Ga–Ga–CD2 moieties (Fig. 4a).
 | ||
| Fig. 4 (a) Products formed when reacting 12+ with a 1:1 mixture of acetonitrile and acetonitrile-d3. (b) Energy landscape and intermediates for a hypothetical mechanism of the C–H bond activation reaction of acetonitrile with 12+ (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures; all values are given in kJ mol−1). The Gibbs free energies were calculated for an oDFB solution (εr = 13.38).32 | ||
Upon heating a 1:1 mixture of separately synthesized [H{Ga(dcpe)}2(CH2CN)]2+ and [D{Ga(dcpe)}2(CD2CN)]2+ for 4 days at 60 °C, no formation of [H{Ga(dcpe)}2(CD2CN)]2+ was observed. Thus, for the reaction of H3C–CN and D3C–CN with [{Ga(dcpe)}2][pf]2, scrambling of Ga–H and Ga–D can be ruled out. This leads to the conclusion that the –H and –CH2CN fragments in the C–H activation product of H3CCN (Scheme 5) are not transferred from the same acetonitrile molecule, i.e., the activation is an intermolecular reaction. A calculated energy landscape of a hypothetical intermolecular reaction mechanism is presented in Fig. 4b, as well as the structures of the corresponding intermediates. The reaction sequence is probably initiated by the coordination of the ligand MeCN to one of the two Ga atoms of the dicationic digallene. The formation of the adduct (B in Fig. 4b) and shift of electron density result in an increased Brønsted basicity of the second Ga atom as well as an increased Brønsted acidity of the methyl group. A subsequent slightly endergonic Brønsted acid–base reaction would yield a tricationic digallane and a zwitterionic, monocationic digallane (C). A second Brønsted acid–base reaction could then lead to isomer D of the observed product E, which is finally formed via a 1,3-rearrangement reaction. The activation barrier for this final reaction step is probably prohibitively high (ca. 135 kJ mol−1), even when taking into account that the reaction requires prolonged heating at 60 °C for 4 days. Thus, it is more likely that the [H2C–CN]− fragment in D is transferred to another gallium dimer in an intermolecular reaction, rather than undergoing a rearrangement reaction. In line with this, no intermediate D is observed spectroscopically during the reaction of 12+ and MeCN, which would be expected if the mechanism presented in Fig. 4b was operative, considering its relative inertness. In conclusion, the calculations underpin that both the C–H bond activation and the [H2C–CN]− migration probably follow a bimolecular mechanism and that (de)protonated MeCN/12+ complexes similar to C and D may be involved.
Overall, the different reactivity of 12+ towards H3C–CN and H2F–CN was unexpected. However, the different routes can be rationalized with quantum chemical calculations: as shown in Scheme 5, the hypothetical [2 + 2] cycloaddition reaction of acetonitrile with 12+ is endergonic in solution, whereas the C–H activation is exergonic, just like the [2 + 2] cycloaddition with the monofluorinated substrate (cf.Scheme 5). These results not only demonstrate that the dicationic digallene shows a versatile chemistry, but also that subtle changes in the substrate can favor different reaction pathways.
 | ||
| Fig. 5 (a) Energy landscape and intermediates for a hypothetical mechanism of the C–F bond activation reaction of 3FB with 12+ (DSD-PBEP86-D3(BJ)/def2-QZVPP(C) single point calculation on RI-BP86(D3BJ)/def2-TZVPP optimized structures); all values are given in kJ mol−1. The Gibbs free energies were calculated for an 3FB solution (εr = 22.1).63 (b + c) Molecular structure in the solid state of the cation in the reaction product of [1][pf]2 with 3FB. The two different perspectives (b) and (c) illustrate the anti-periplanar arrangement of the F atom and the C6F2H3 moiety in the product. Only one of the three crystallographically independent [F{Ga(dcpe)}2C6F2H3]2+ units is shown. Hydrogen atoms, counteranions and disordered ligand molecules are omitted for clarity; the cyclohexyl groups are depicted in wireframe for simplicity. Thermal ellipsoids are set at the 50% probability level. | ||
Starting from the complex B, the F atom and the C6F2H3 moiety are transferred to the two Ga atoms of the digallene in a concerted fashion. The Ga–C and Ga–F bonds are formed and the C–F bond is broken in the transition state TSB/C, which is ca. 100 kJ mol−1 higher in energy than B. Subsequently, a thermodynamically favorable rotation around the Ga–Ga bond in the initially formed syn-periplanar product C diminishes the steric repulsion between the cyclohexyl groups and yields the anti-periplanar product D in the crystallized compound.
A comparison of the reactivity of 12+ towards fluorinated benzenes reveals that the reactivity of the digallene is pronounced towards 3FB, while the reactions with both higher and lower fluorinated aromatics are considerably slower. Solutions of [1][pf]2 in oDFB are stable over several days to weeks at rt before the (very) slow formation of Ga–F bonds, which can be observed via NMR spectroscopy. By contrast, even at 60 °C, the digallene is virtually unreactive towards hexafluorobenzene (6FB) (Scheme 6), even though the C–F bonds in 6FB are easily cleaved with a bimetallic Mg(I) complex64 and a neutral alumene.65,66 It should be mentioned that 12+ is similarly reactive towards 1,2,3,4-tetrafluorobenzene (4FB) as towards 3FB. However, the reaction is considerably less selective, as multiple C–F bond activation products are observed spectroscopically after 3 days at 40 °C (Section 4.16.2 in the SI).
Different and opposing effects may contribute to the observed reactivity trend: on the one hand, it is well known that Caromatic–F bonds are weakened by additional fluorine substituents, accounting for the fact that C–F bonds in higher fluorinated aromatics are usually easier to activate than in lower fluorinated aromatics.58–60 This was experimentally confirmed with main group element compounds, e.g. a neutral alumene65,66 and a neutral, dinuclear bisgallene.22 On the other hand, the coordinating ability decreases with a higher degree of fluorination. It may also be significant that the steric demand of the aromatic substrate increases with every F substituent and that the polarity of the fluorinated benzenes reaches a maximum with 3FB with a large dipole moment of 4.7 Debye,63 before decreasing with increasing degree of fluorination.63 Thus, the reaction outcomes with fluorinated arenes nicely illustrate that the reactivity of 12+ towards fluorinated compounds is influenced by the strength and polarity of the C–F bond, as well as the coordinating ability of the substrate, which in turn is affected by electronic and steric effects. These partly opposing effects lead to the observed high reactivity and selective reaction of 12+ towards 3FB, i.e., a polar arene with a medium degree of fluorination. It is noteworthy that the reaction between a neutral alumene and 3FB yields both conceivable C–F activation products and requires prolonged heating at 80 °C for several days,66 which is in contrast to the selective, facile bond cleavage with 12+ at 40 °C.
Overall, the results imply that C–F bond activation with dicationic digallenes is possible, if the substrate has an additional “donor” functionality or a reasonably electron rich π bond or aromatic π system. Interestingly, in the case of fluorinated aromatics, the dicationic digallene shows a different reactivity trend than neutral subvalent group 13 compounds, suggesting that the cationic congeners may complement the reactivity of the neutral agents.
Conclusion
Herein, the reactivity of a dicationic digallene towards CC multiple bonds and very strong element–element E–Y single bonds was thoroughly investigated. The instantaneously formed, strictly dimeric [{Ga(dcpe)}2][pf]2 with a trans-bent double bond served as a model compound, as summarized in Scheme 10.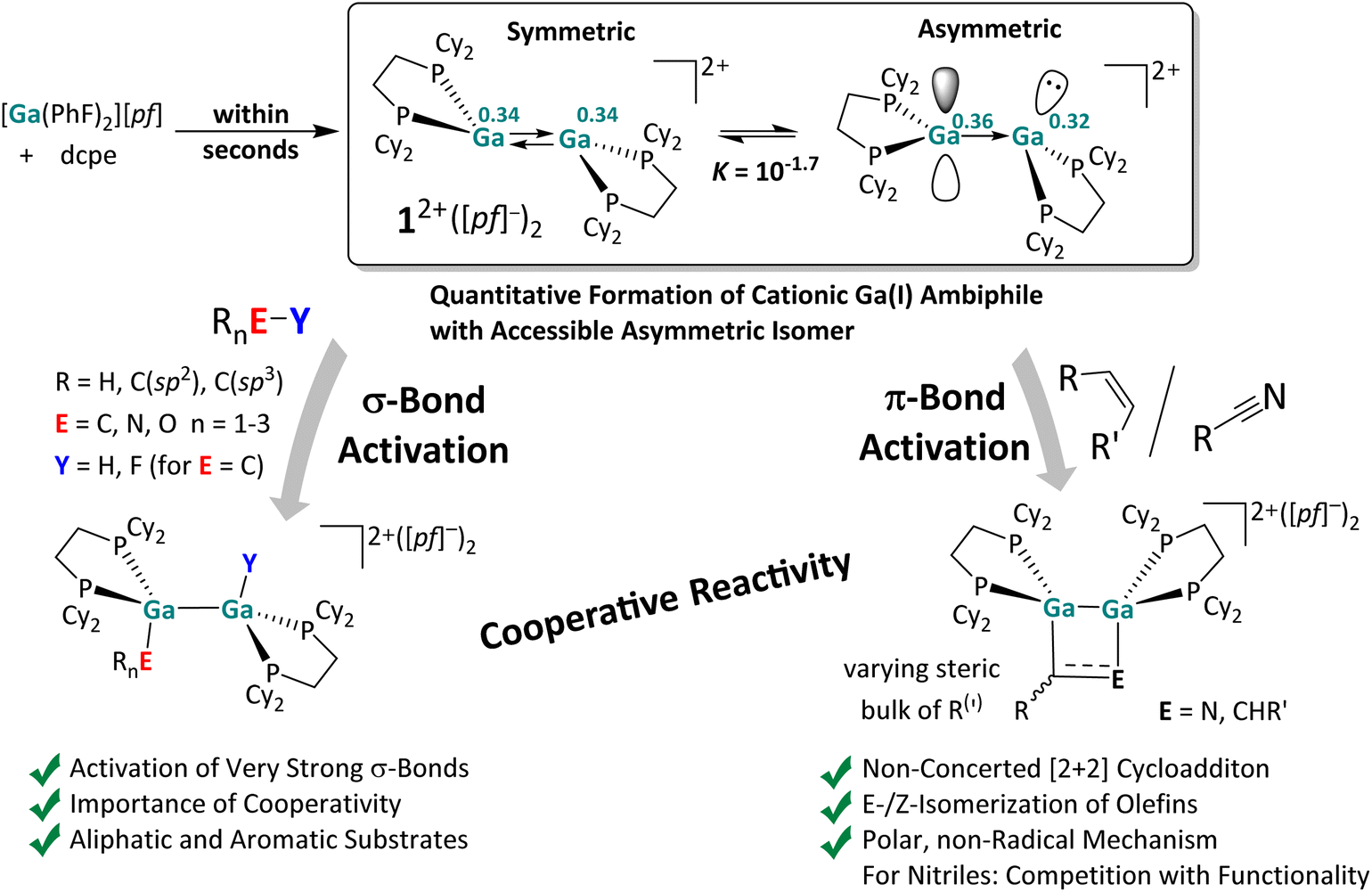 | ||
| Scheme 10 Results of this work. The in situ formed reactive digallene 12+ accessible from [Ga(PhF)2][pf] and commercial dcpe,29 reacts cooperatively and with strong contributions from the accessible asymmetric conformer (QTAIM charges on Ga atoms are written in superscript) with olefins, nitriles and strong C–F, C–H, N–H, and O–H σ-bonds. The herein derived mechanistic insight will help to pick up selected activations and to transform them into parts of catalytic cycles, especially in those cases, where the energetics underlying the activations is only moderately exergonic or even on the edge (olefins, N–H, O–H, and CN-cycloaddition). | ||
To the best of our knowledge, our results include the first additions of H–O and H–C bonds to a trans-bent ditrielene double bond and the first C–F bond activation with a subvalent cationic group 13 complex. Notably, the [2 + 2] cycloaddition reaction between a dicationic digallene and an alkene is a stepwise reaction and most likely a polar addition, without radical intermediates. The starting materials, cycloaddition products and open-chain intermediates are in equilibrium with each other, with the steric bulk of the substrate determining the position of the equilibrium. Consequently, employing doubly substituted cis-alkenes does not yield the expected cycloaddition product, but instead cis/trans mixtures of the starting material, which may also be obtained catalytically.
In addition, we demonstrated the bond cleavage of very strong single bonds, i.e., H–O, H–N, H–C and F–C bonds with [{Ga(dcpe)}2]2+, under mild conditions. The H–O and H–N bonds are even cleaved under ambient conditions. These results underline the pronounced reactivity of dicationic digallenes – probably proceeding through an accessible asymmetric conformer, which was shown to be only 10 kJ mol−1 higher in energy than the ground state using DFT calculations. The electronic influence of substrate/digallene adduct formation is clearly visible from the QTAIM and EDA-NOCV analyses, revealing asymmetric charge redistribution facilitating the bond activation. Quantum chemical calculations indicate that the H–N, H–O and C–F bonds are likely cleaved in a concerted, cooperative mechanism, promoted by substrate coordination. The oxidative addition into the H–C bond of acetonitrile probably also proceeds via initial coordination of MeCN and a series of protonation/deprotonation steps. It appears that coordination of the substrate to the Ga⇄Ga double bond is a prerequisite for activation of strong bonds, as similar observations were made with fluorinated organic compounds: while [{Ga(dcpe)}2]2+ cleaves the C–F bonds in substrates with reasonably electron rich aromatic π-systems or double bonds in a highly exergonic reaction, it does not react with very poor ligands. Notably, the facile C–F cleavage in 3FB, which is difficult to activate with neutral, subvalent group 13 species, implies that the positive charge of the digallene may induce a reactivity, which is complementary to that of the neutral congeners.
These results shall spark the application of cationic digallenes as in situ accessible catalysts in organic reactions, very much like in classical transition metal chemistry. To this end, further reversible bond activation reactions have to be achieved. Olefin/digallene or nitrile/digallene combinations are the most promising candidates for a catalytic cycle, in light of the reversibility of the [2 + 2] cycloaddition. While the thermodynamic stability of the C–F activation products probably impedes follow-up reactions, the N–H and O–H addition products are less thermodynamically stable and may thus serve as starting points for catalytic reactions. Further experiments are under way in order to fine tune the experimental conditions, find a suitable ligand/substrate combination, and to eventually perform main group metal catalysis with subvalent gallium.
Author contributions
A. B., N. G. K. and I. K. conceived the experiments. N. G. K., A. B., and C. R. performed the experiments. H. S. measured the NMR spectra. All authors analyzed and discussed the experimental data. A. B. and I. K. co-wrote the paper and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): experimental details, procedures, weights, and IR- and NMR spectra of the reactions; details of the quantum chemical calculations together with crystallographic details. See DOI: https://doi.org/10.1039/d5sc09508e.CCDC 2488257–2488261 and 2490061 contain the supplementary crystallographic data for this paper.69a–f
Acknowledgements
This work was supported by the Albert-Ludwigs-Universität Freiburg and by the German Research Foundation (DFG; project number: 400773200). I. K. acknowledges the support through the European Research Council's ERC Advanced Grant InnoChem (Grant agreement ID: 101052935). The authors acknowledge support from the state of Baden-Württemberg through bwHPC and the DFG through grant numbers INST 40/467-1 and 575-1 FUGG (JUSTUS1 and 2 cluster). We would like to thank Dr Harald Scherer, Celine Schullian and MSc. Tanja Oppermann for the measurement of the NMR spectra and Dr Burkhard Butschke for his support regarding single crystal X-ray crystallography. A. B. thanks the “Fonds der Chemischen Industrie” and C. R. thanks the “Jürgen Manchot Stiftung” for PhD fellowships.Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- R. L. Melen, Science, 2019, 363, 479–484 Search PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- J. P. Malrieu and G. Trinquier, J. Am. Chem. Soc., 1989, 111, 5916–5921 Search PubMed.
- (a) G. Trinquier, J. P. Malrieu and P. Riviere, J. Am. Chem. Soc., 1982, 104, 4529–4533 CrossRef CAS; (b) E. A. Carter and W. A. Goddard, J. Phys. Chem., 1986, 90, 998–1001 Search PubMed.
- P. P. Power, Chem. Rev., 1999, 99, 3463–3504 CrossRef CAS PubMed.
- P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 317 RSC.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 Search PubMed.
- (a) J. T. Snow, S. Murakami, S. Masamune and D. J. Williams, Tetrahedron Lett., 1984, 25, 4191–4194 CrossRef CAS; (b) S. Collins, S. Murakami, J. T. Snow and S. Masamune, Tetrahedron Lett., 1985, 26, 1281–1284 CrossRef CAS.
- (a) D. E. Goldberg, D. H. Harris, M. F. Lappert and K. M. Thomas, J. Chem. Soc., Chem. Commun., 1976, 261–262 RSC; (b) C. R. Stennett, M. Bursch, J. C. Fettinger, S. Grimme and P. P. Power, J. Am. Chem. Soc., 2021, 143, 21478–21483 CrossRef CAS PubMed.
- (a) M. Stürmann, M. Weidenbruch, K. W. Klinkhammer, F. Lissner and H. Marsmann, Organometallics, 1998, 17, 4425–4428 Search PubMed; (b) M. Stürmann, W. Saak, H. Marsmann and M. Weidenbruch, Angew. Chem., Int. Ed., 1999, 38, 187–189 Search PubMed.
- (a) S. A. Batcheller and S. Masamune, Tetrahedron Lett., 1988, 29, 3383–3384 CrossRef CAS; (b) D. Wendel, T. Szilvási, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 9156–9159 Search PubMed; (c) F. Hanusch, L. Groll and S. Inoue, Chem. Sci., 2020, 12, 2001–2015 Search PubMed.
- D. Wendel, T. Szilvási, D. Henschel, P. J. Altmann, C. Jandl, S. Inoue and B. Rieger, Angew. Chem., Int. Ed., 2018, 57, 14575–14579 Search PubMed.
- N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2002, 41, 2842–2844 CrossRef CAS PubMed.
- (a) N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 2667–2679 CrossRef CAS PubMed; (b) R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 Search PubMed; (c) M. L. McCrea-Hendrick, C. A. Caputo, C. J. Roberts, J. C. Fettinger, H. M. Tuononen and P. P. Power, Organometallics, 2016, 35, 579–586 Search PubMed; (d) A. Lehmann, J. D. Queen, C. J. Roberts, K. Rissanen, H. M. Tuononen and P. P. Power, Angew. Chem., Int. Ed., 2024, 63, e202412599 CrossRef CAS PubMed; (e) R. J. Schwamm, M. A. Bhide, G. S. Nichol and M. J. Cowley, Chem. Sci., 2025, 16, 13333–13344 RSC.
- Á. García-Romero, I. Fernández and J. M. Goicoechea, Angew. Chem., Int. Ed., 2025, 64, e202509661 CrossRef PubMed.
- X. Liu, S. Dong, J. Zhu and S. Inoue, J. Am. Chem. Soc., 2024, 146, 23591–23597 CrossRef CAS PubMed.
- Z. Zhu, X. Wang, Y. Peng, H. Lei, J. C. Fettinger, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2009, 48, 2031–2034 Search PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC.
- (a) E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9758–9759 CrossRef CAS; (b) R. J. Schwamm, M. D. Anker, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2018, 57, 5885–5887 CrossRef CAS; (c) R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS.
- O. Kysliak, H. Görls and R. Kretschmer, J. Am. Chem. Soc., 2021, 143, 142–148 Search PubMed.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- M. Driess and H. Grützmacher, Angew. Chem., Int. Ed., 1996, 35, 828–856 CrossRef CAS.
- A. Barthélemy, H. Scherer, M. Daub, A. Bugnet and I. Krossing, Angew. Chem., Int. Ed., 2023, 62, e202311648 Search PubMed.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- B. Ruscic, D. Feller and K. A. Peterson, Theor. Chem. Acc., 2014, 133, 1415 Search PubMed.
- C. Wohlfarth and Y.-R. Luo, in CRC handbook of chemistry and physics, ed. W. M. Haynes, D. R. Lide, T. J. Bruno, and W. M. Haynes, CRC Press, Boca Raton, 97th edn, 2017, 6-199–6-220 and 9-73–6-86 Search PubMed.
- A. Barthélemy, P. Dabringhaus, E. Jacob, H. Koger, D. Röhner, M. Schmitt, M. Sellin, and I. Krossing , in Comprehensive Inorganic Chemistry III, ed. J. Reedijk, and K. R. Poeppelmeier, Elsevier, Amsterdam, 3rd edn, 2023, pp. 378–438 Search PubMed.
- (a) R. J. Wehmschulte, R. Peverati and D. R. Powell, Inorg. Chem., 2019, 58, 12441–12445 CrossRef CAS PubMed; (b) E. Herappe-Mejía, K. Trujillo-Hernández, J. Carlos Garduño-Jiménez, F. Cortés-Guzmán, D. Martínez-Otero and V. Jancik, Dalton Trans., 2015, 44, 16894–16902 RSC; (c) M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC.
- P. Dabringhaus, T. Heizmann and I. Krossing, Chem.–Eur. J., 2023, 29, e202302212 CrossRef CAS.
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem.–Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed.
- A. Barthélemy and I. Krossing, Inorg. Chem., 2024, 63, 21763–21787 CrossRef.
- P. Dabringhaus, A. Barthélemy and I. Krossing, Z. Anorg. Allg. Chem., 2021, 647, 1660–1673 CrossRef CAS.
- M. Schorpp, R. Tamim and I. Krossing, Dalton Trans., 2021, 50, 15103–15110 RSC.
- P. Dabringhaus, H. Scherer and I. Krossing, Nat. Synth., 2024, 3, 732–743 CrossRef CAS.
- J. M. Slattery, A. Higelin, T. Bayer and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 3228–3231 CrossRef CAS PubMed.
- (a) I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS; (b) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC; (c) I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- (a) K. Glootz, D. Kratzert, D. Himmel, A. Kastro, Z. Yassine, T. Findeisen and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 14203–14206 CrossRef CAS PubMed; (b) K. Glootz, D. Himmel, D. Kratzert, B. Butschke, H. Scherer and I. Krossing, Angew. Chem., Int. Ed., 2019, 58, 14162–14166 CrossRef CAS PubMed; (c) A. Barthélemy, H. Scherer and I. Krossing, Chem.–Eur. J., 2022, 28, e202201369 CrossRef PubMed.
- (a) L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC; (b) J. M. L. Martin and G. Santra, Isr. J. Chem., 2020, 60, 787–804 CrossRef CAS.
- H. N. Dunning, Ind. Eng. Chem., 1953, 45, 551–564 CrossRef CAS.
- (a) R. G. Bergman and S. A. Sherrod, J. Am. Chem. Soc., 1971, 93, 1925–1940 CrossRef CAS; (b) S. E. Gottschling, T. N. Grant, K. K. Milnes, M. C. Jennings and K. M. Baines, J. Org. Chem., 2005, 70, 2686–2695 CrossRef CAS PubMed.
- L. F. Tietze, and G. Kettschau, in Topics in Current Chemistry, ed. A. de Meijere, K. N. Houk, J.-M. Lehn, S. V. Ley, J. Thiem, B. M. Trost, F. Vögtle, H. Yamamoto, and P. Metz, Springer, Berlin, Heidelberg, 1997, vol. 189, pp. 1–120 Search PubMed.
- A. Barthélemy, Investigations of the Ligand-Dependent Clustering Tendency and Reactivity of Cationic, Subvalent Gallium and Indium Complexes, PhD thesis, Albert-Ludwigs University Freiburg, 2024 Search PubMed.
- C. A. Caputo, J.-D. Guo, S. Nagase, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2012, 134, 7155–7164 CrossRef CAS PubMed.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- S. L. McOnie, G. A. Özpınar, J. L. Bourque, T. Müller and K. M. Baines, Dalton Trans., 2021, 50, 17734–17750 RSC.
- D. Sarkar, P. Vasko, L. Ying, J. J. C. Struijs, L. P. Griffin and S. Aldridge, Angew. Chem., Int. Ed., 2025, 64, e202502326 CrossRef CAS PubMed.
- A. Barthélemy, H. Scherer, H. Weller and I. Krossing, Chem.–Eur. J., 2024, 30, e202400897 CrossRef PubMed.
- (a) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (b) K. I. Goldberg, A. S. Goldman, in Organometallic C-H Bond Activation: An Introduction, ed. K. I. Goldberg and A. S. Goldman, ACS symposium series, American Chemical Society, Washington DC, 2004, vol. 885, pp. 1–43 Search PubMed; (c) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 Search PubMed.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376–20380 CrossRef CAS PubMed.
- C. Jones, D. P. Mills and R. P. Rose, J. Organomet. Chem., 2006, 691, 3060–3064 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- S. A. Johnson, J. A. Hatnean, and M. E. Doster in Progress in Inorganic Chemistry, ed. K. D. Karlin, Wiley, 2011, 255–352 Search PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- S. A. Macgregor, D. McKay, J. A. Panetier and M. K. Whittlesey, Dalton Trans., 2013, 42, 7386–7395 RSC.
- O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed.
- (a) R. L. Luck and R. H. Morris, J. Am. Chem. Soc., 1984, 106, 7978–7979 Search PubMed; (b) E. Becker, C. Slugovc, E. Rüba, C. Standfest-Hauser, K. Mereiter, R. Schmid and K. Kirchner, J. Organomet. Chem., 2002, 649, 55–63 CrossRef CAS; (c) Q. Teng, P. S. Ng, J. N. Leung and H. V. Huynh, Chem.–Eur. J., 2019, 25, 13956–13963 Search PubMed; (d) N. G. Kub, R. Sievers, J. Parche and M. Malischewski, Chem.–Eur. J., 2024, 30, e202400427 CrossRef CAS PubMed.
- C. Armbruster, M. Sellin, M. Seiler, T. Würz, F. Oesten, M. Schmucker, T. Sterbak, J. Fischer, V. Radtke and J. Hunger, et al. , Nat. Commun., 2024, 15, 6721 CrossRef CAS PubMed.
- C. Bakewell, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2016, 138, 12763–12766 CrossRef CAS PubMed.
- M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 RSC.
- T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363–5365 Search PubMed.
- A. K. Hijazi, A. Al Hmaideen, S. Syukri, N. Radhakrishnan, E. Herdtweck, B. Voit and F. E. Kühn, Eur. J. Inorg. Chem., 2008, 2008, 2892–2898 Search PubMed.
- S. S. Andrews and S. G. Boxer, J. Phys. Chem. A, 2000, 104, 11853–11863 CrossRef CAS.
- (a) CCDC 2488257: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pj7cv; (b) CCDC 2488258: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pj7dw; (c) CCDC 2488259: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pj7fx; (d) CCDC 2488260: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pj7gy; (e) CCDC 2488261: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pj7hz; (f) CCDC 2490061: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pl3kz.
Footnotes |
| † The previously calculated thermodynamic values for the dissociation of 12+ in oDFB (ΔrH° ≈ +105 kJ mol−1 and ΔrG° ≈ +43 kJ mol−1 at 298 K)29 indicate that the complex largely remains dimeric in solution, not only at rt, but also at slightly elevated temperatures, e.g. at 60 °C. In line with this, diluting or heating the sample at 60 °C does not lead to a significant colour change of the dark red solution. By contrast, monocationic, monomeric gallenes are reported to produce yellow solutions.40 Therefore, in order to play a role in the bond activation reactions, the monomer, which may be present in extremely small quantities, would have to be considerably more reactive than the dimer. This contradicts not only the FMO analysis (Fig. 1) but also our experience with similar, cationic (di)gallene systems.40 Finally, the most important observation is that the reactions of 12+ and one equivalent of the substrate E–Y yield exclusively dimeric products, as confirmed by NMR spectroscopy and scXRD. As demonstrated previously, monomeric GaIII addition products can be obtained, but only with a second equivalent of the substrate E–Y and via the dimeric GaII addition products.29 In the present work, a monomeric intermediate was only observed with a slight surplus of HNPh2, but already at rt, implying that the bulky amine partially breaks down the dimer (Scheme 9). These results strongly suggest that no monomeric species are involved in the bond activation reactions presented herein and that the dimeric 12+ acts as the active species in solution. |
‡ Note that the CN stretching mode ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CN in the C–H activation product of H3CCN and in the C–F activation product of FH2CCN is red-shifted (ca. 2222 cm−1, see Scheme 5) compared to free H3CCN (2253 and 2293 cm−1).67 For both insertion products, the vibrational Stark effect, i.e. an electric field effect of the nitrile substituents, should account for the weakening of the CN triple bond.68 CN in the C–H activation product of H3CCN and in the C–F activation product of FH2CCN is red-shifted (ca. 2222 cm−1, see Scheme 5) compared to free H3CCN (2253 and 2293 cm−1).67 For both insertion products, the vibrational Stark effect, i.e. an electric field effect of the nitrile substituents, should account for the weakening of the CN triple bond.68 |
| This journal is © The Royal Society of Chemistry 2026 |