DOI:
10.1039/D5SC09144F
(Edge Article)
Chem. Sci., 2026,
17, 4942-4947
Self-induced charge transfer activation enables metal-free C–H coupling of polycyclic aromatic hydrocarbons under photo irradiation
Received
23rd November 2025
, Accepted 6th February 2026
First published on 10th February 2026
Abstract
Axially chiral three-dimensional nanographenes (3D NGs) represent promising scaffolds for chiral optoelectronic materials, yet their direct synthesis remains challenging because of the oxidative fragility of π-extended arenes. Here we report a metal-free, photo-irradiated oxidative C–H/C–H biaryl coupling of polycyclic aromatic hydrocarbons mediated by a Brønsted acid and O2. The reaction efficiently converts π-extended arenols, including fluoranthene derivatives, into structurally diverse axially chiral 3D NGs without overoxidation. Mechanistic studies reveal that self-induced charge transfer (CT) complexation between protonated and neutral arenols triggers photoinduced formation of radical cations, as supported by UV-vis, ESR and DFT analyses. The obtained axially chiral 3D NGs exhibit high configurational stability and photophysical features rationalized by TD-DFT calculations. This strategy establishes a general platform for constructing axially chiral 3D NGs from π-extended arenols under metal-free conditions.
Introduction
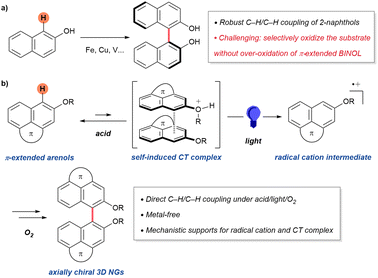
Nanographenes (NGs), atomically precise graphene fragments with tunable dimensionally, have emerged as ideal scaffolds for chiral optoelectronic systems.1 In particular, three-dimensional (3D) NGs are attracting attention because their contoured, shape-persistent topologies mitigate π–π aggregation while amplifying chiroptical responses. Representative architectures based on triptycene-,2,3 cyclooctatetraene-,4–7 spirocyclic-,8,9 and biaryl10–15-frameworks (Fig. 1a) exemplify this design principle. Among these, biaryl-type NGs are attractive for accessing axially chiral frameworks and serving as precursors to more complex 2D and 3D materials.8,16,17 However, only a limited handful of synthetic examples have been reported (Fig. 1b). Because most reported routes rely on cross couplings of prefunctionalized substrates, more direct strategies would expand the accessible chemical space of biaryl-type NGs and accelerate materials discovery.
 |
| | Fig. 1 (a) Representative core scaffolds of reported 3D NGs. Polycyclic aromatic hydrocarbon moiety (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ar) is simplified and shown. (b) Reported axially chiral 3D NGs and their synthetic strategies. R = tert-butyl, Ar1 = 2,4,6-trimethylphenyl. Ar) is simplified and shown. (b) Reported axially chiral 3D NGs and their synthetic strategies. R = tert-butyl, Ar1 = 2,4,6-trimethylphenyl. | |
A direct C–H coupling would represent the most straightforward route to biaryl-type NGs. While transition-metal-catalyzed,18,19 photoredox-catalyzed,20,21 or hypervalent iodine22,23-mediated biaryl couplings of simpler arenes, such as benzene and naphthalene derivatives, are known, these methods tend to be less effective for π-extended PAHs, where multiple reactive sites and oxidative instability result in poor regioselectivity or even decomposition.18,24,25 Classical BINOL synthesis elegantly exploits the hydroxyl group to direct coupling at the C1 position of 2-naphthol, yet analogous strategies have rarely succeeded in π-extended systems due to overoxidation of the biaryl product (Fig. 2a).16,26–31 Despite recent advances in π-extended systems, the synthesis of axially chiral 3D NGs via Scholl-type C–H coupling remains a major challenge and has been only rarely achieved.13,15
 |
| | Fig. 2 (a) Classical approach to synthesize BINOL. (b) Metal-free C–H/C–H biaryl coupling of π-extended arenols enabled by self-induced CT complex under acid/light/O2 condition (this work). | |
To overcome this limitation, we envisioned a photo-irradiated self-activation mechanism: an in situ transient protonated π-extended arenol with its neutral one generates a self-induced pseudo-homo charge transfer (CT) complex, which upon photoexcitation produces a radical cation competent for C–H coupling. Unlike the persistent hetero CT complexes between separate electron donors and acceptors reported by Sanford32,33 and Kozlowski,34,35 our system relies on intramolecular charge polarization within a single arenol scaffold. This design allows oxidative coupling of large, π-extended substrates while suppressing overoxidation through steric and electronic self-regulation. Specifically, because the targeted products are 3D NGs, their nonplanar geometry imposes substantial steric hindrance that prevents close π–π interaction between arenol units, making CT complexation less favorable. As a result, overoxidation of the resulting biaryl products, a common issue in π-extended systems, is mitigated. Herein, we report an acid, light and O2 mediated C–H/C–H biaryl coupling, elucidate its mechanism, and disclose the properties of axially chiral 3D NGs.
Results and discussion
Optimization
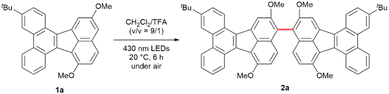
During the course of our study on non-benzenoid PAHs,36–38 we commenced our study with the oxidative biaryl coupling of dibenzo[j,l]fluoranthene 1a as a model substrate, followed by optimization of the reaction conditions (Table 1). Following a comprehensive evaluation of reaction parameters, we found that 430 nm LEDs irradiation of a solution of 1a in CH2Cl2/TFA (v/v = 9/1) under air for 6 h at 20 °C afforded the desired biaryl product 2a in 90% yield (entry 1). Notably, 2a was obtained as a single regioisomer, and this regioselectivity is supported by DFT calculations (see Fig. S26). No conversion was observed in the absence of TFA (entry 2), underscoring the crucial role of Brønsted acid in this transformation. Replacement of TFA (pKa = −0.3) with weaker acid such as acetic acid (AcOH, pKa = +4.8) resulted in full recovery of 1a (entry 3). Conversely, the use of a stronger acid, methanesulfonic acid (MsOH, pKa = −2.6) restored high efficiency, affording 2a in 90% yield (entry 4). The reaction proved highly sensitive to the atmosphere: under N2, the yield plummeted, indicating that O2 serves as a terminal oxidant (entry 5). In the dark, performing the reaction led to only trace amounts of product (entry 6), confirming the indispensable role of photoirradiation. Taken together, these results indicate that the combination of Brønsted acid, O2, and light is essential for this oxidative biaryl coupling reaction. Notably, applying known BINOL coupling conditions, such as FeCl3 (entry 7) or DDQ (entry 8) as oxidants (representative examples; see Table S5), to 1a led to negligible conversion or severe overoxidation, resulting in poor mass balance.
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Deviation |
Yieldsb (%) |
|
2a
|
1a (recov.) |
|
Reaction conditions: 1a (0.030 mmol) in CH2Cl2/TFA (v/v = 9/1, 0.030 M) at 20 °C for 6 h, irradiating with 430 nm LEDs.
Yields were determined by 1H NMR using triphenylmethane as an internal standard. Isolated yield was shown in parentheses.
Reaction conditions: 1a (0.050 mmol) and FeCl3 (0.20 mmol) in EtOH (1.4 mL) at 95 °C (reflux) for 15 h.
Reaction conditions: 1a (0.050 mmol), DDQ (0.10 mmol) and TfOH (0.30 mmol) in CH2Cl2 (5.0 mL) at 0 °C for 10 min.
|
|
1
|
None
|
90 (85)
|
0 |
| 2 |
No TFA |
0 |
86 |
| 3 |
AcOH instead of TFA |
0 |
93 |
| 4 |
MsOH instead of TFA |
90 |
0 |
| 5 |
N2 instead of air |
2 |
91 |
| 6 |
In dark |
7 |
79 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
Known conditions
|
| 7c |
FeCl3 in EtOH, reflux |
(8) |
(87) |
| 8d |
DDQ and TfOH in CH2Cl2, 0 °C |
21 |
3 |
Substrate scope
With the optimized reaction conditions in hand, we next investigated the substrate scope of fluoranthene derivatives 1 in dimerization reaction (Fig. 3). As a preliminary investigation, we evaluated the effect of varying the C1 substituent on a 5-methoxydibenzo[j,l]fluoranthene scaffold (1a–1d). Electron donating groups at C1 (1a, 1b) and the unsubstituted parent substrate (1c) were well tolerated in this reaction, giving corresponding axially chiral 3D NGs 2a–2c in high yield (85%, 99% and 75%, respectively). In contrast, introduction of an electron withdrawing group at C1 (1d) led to a markedly lower yield (28%), indicating that high electron density of an aromatic ring is essential for efficient dimerization. Next, we used 5-hydroxydibenzo[j,l]fluoranthene (1e) as a substrate, and the dimerization proceeded in a moderate yield (64%). We attribute the slight decrease in yield compared to 1a–1c to the instability of 1e, which is prone to oxidation under air. Finally, 5-methoxyfluoranthene (1f) afforded a high yield (74%) of 2f, albeit that required replacement of TFA with MsOH.
 |
| | Fig. 3 Substrate scope for fluoranthene derivatives 1a. aReaction conditions: 1 (0.030 mmol) in CH2Cl2/TFA (v/v = 9/1, 0.030 M) at 20 °C. Isolated yields are shown. Wavelength of LEDs and reaction time are indicated in parentheses. b0.20 mmol of 1f and MsOH instead of TFA were conducted. | |
Furthermore, to delineate the substrate scope beyond the fluoranthene scaffold, we investigated the dimerization reaction using other arenols (Fig. 4). Reaction parameters (irradiation wavelength, reaction temperature and time) were tailored to each substrate, and the addition of MS3A provided a modest increase in yield (see Table S6–S14). With 2-pyrenol (3a), the corresponding dimer 4a was isolated in 82% yield and its structure was confirmed by X-ray crystallographic analysis. 9-Phenanthrenol (3b) and 2-anthracenol (3c) likewise furnished the corresponding dimers (76% and 48%, respectively). Notably, this dimerization reaction using 3b can be performed on a 1.0 mmol scale without loss in efficiency (85%). In the naphthalene skeletons, 2-hydroxynaphthalene (3d) gave BINOL in 70% yield, whereas 2-methoxynaphthalene (3e) afforded only 12% yield with 70% recovery of 3e; this difference is consistent with the lower propensity of 3e to form the CT complex under these conditions (see Fig. S14 and S18). 1,4-Bis(2-naphthoxy)butane (3f) did not deliver the intramolecular product 4f, likely because the tether restricted conformational freedom and disfavored formation of CT complex. In addition, 2,3- and 2,7-dihydroxynaphthalene (3g, 3h) reacted smoothly, and 2,6-dihydroxynaphthalene (3i) furnished 4i in high yield (72%). Using 2-hydroxy-6-methoxynaphthalene (3j) gave two regioisomers, 4j (C1–C1′) and 4j′ (C1–C5′), in a 7:1 ratio (62% yield). Consistent with the result for 3e, 2,6-dimethoxynaphthalene (3k) afforded a lower yield (25%) of dimer 4k. Collectively, these results showed that our dimerization protocol was effective not only for the fluoranthene scaffold but also for several other PAHs.
 |
| | Fig. 4 Substrate scope. aReaction conditions: 3 (0.40 mmol) and MS3A (10 wt%) in CH2Cl2/TFA (v/v = 1/1, 0.10 M). Wavelength of LEDs, reaction temperature and time are indicated in parentheses. Isolated yields are shown. b0.20 mmol of 3a. cOn 1.0 mmol scale. d0.060 mmol of 3f in CH2Cl2/TFA (v/v = 9/1, 0.015 M). e0.12 mmol of substrate in CH2Cl2/TFA (v/v = 9/1, 0.030 M). fWithout MS3A. | |
Mechanistic studies
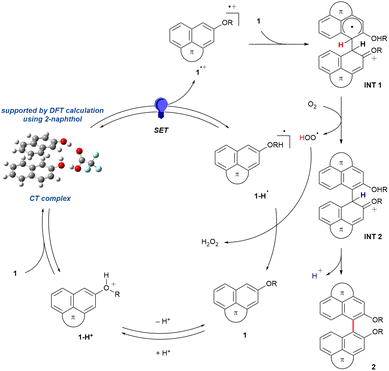
To gain insight into the reaction mechanism, we first recorded the UV-vis absorption spectra of 1a (Fig. 5a). In CH2Cl2, no significant absorption was observed above 550 nm. However, in CH2Cl2/TFA (v/v = 9/1), its UV-vis spectrum displayed a new broad absorption band with a maximum at ca. 640 nm and a tail extending into the near-infrared (NIR) region. This spectral change suggests the formation of a new species under acidic conditions. The absorption at 640 nm was further enhanced upon photoirradiation. To further investigate the reaction mechanism, we conducted the biaryl coupling reaction in the presence of TEMPO (5.0 eq.), a well-known radical scavenger (Fig. 5b). In this case, the reaction gave a complex mixture, suggesting the involvement of radical intermediates. To further support the formation of radical species, ESR spectroscopy was carried out (Fig. 5c). While no ESR signal was observed in CH2Cl2 alone, an ESR signal (g = 2.0045) appeared in CH2Cl2/TFA (v/v = 9/1), consistent with the formation of a typical radical species.39,40 Moreover, photoirradiation led to a further increase in ESR signal intensity, indicating that light promotes the generation of the radical species. Finally, we employed tris(4-bromophenyl)ammoniumyl hexachloroantimonate (Magic Blue) as an external single electron oxidant without TFA and light irradiation (Fig. 5d). As a result, the biaryl coupling reaction proceeded smoothly, indicating that a radical cation intermediate 1a˙+ is involved in the reaction pathway. UV-vis titration of 1a with Magic Blue reproduced the ca. 640 nm band and TD-DFT calculations assign this transition to 1a˙+ (see Fig. S12, S25 and Table S28).
 |
| | Fig. 5 Mechanistic studies. (a) UV-vis absorption and (c) ESR spectra of 1a (30 mM). Black line: in CH2Cl2, blue line: in CH2Cl2/TFA (v/v = 9/1), red line: in CH2Cl2/TFA (v/v = 9/1) upon irradiation with 430 nm LEDs for 5 min. (b) Biaryl coupling with TEMPO as a radical scavenger. (d) Biaryl coupling with Magic Blue as a single electron oxidant. | |
Proposed mechanism
Based on our observations, we propose the reaction mechanism depicted in Scheme 1. A self-induced pseudo-homo CT complex is first formed between neutral fluoranthene 1 and its protonated counterpart (1-H+). Geometry optimization using 2-naphthol as a simplified model supports the feasibility of such an association, and subsequent natural transition orbital (NTO) analysis41 indicates that photoexcitation promotes intramolecular charge transfer leading to a radical cation species (see Fig. S17). The radical cation (1˙+) subsequently undergoes coupling with 1 to form intermediate INT 1.42,43 Then, O2 formally abstracts a hydrogen atom from INT 1 to give INT 2, while the resulting HOO˙ abstracts another hydrogen atom from 1-H˙, thereby generating H2O2 and simultaneously regenerating 1.32,34,35 Finally, deprotonation of INT 2 furnishes the axially chiral 3D NG 2.
 |
| | Scheme 1 Proposed mechanism. Fluoranthene 1 is shown in a simplified form for clarity (R = Me or H). | |
Physical properties
To further investigate the chiroptical properties of the coupling products, we subjected rac-2a to chiral HPLC, which clearly separated the two enantiomers ((+)-2a and (−)-2a) that exhibited mirror-image CD spectra (Fig. 6a). Comparison of the experimental CD spectra with TD-DFT calculations enabled assignment of (−)-2a as the R enantiomer. Theoretical calculations revealed a high racemization barrier of the model structure (ΔG‡calc = 36.5 kcal mol−1), which is comparable to that of typical BINOL derivatives.44 This theoretically estimated racemization barrier is in good agreement with the experimentally estimated one (ΔG‡exp = 36.9 kcal mol−1), supporting the calculated racemization mechanism (see Fig. S27 and S28). Finally, the photophysical properties of fluoranthene 1a and axially chiral 3D NG 2a were compared (Fig. 6b). In the UV-vis absorption spectrum, 1a displayed pronounced absorption bands at 418 nm (ε = 1.28 × 104 L mol−1 cm−1) and a broad feature spanning 430–500 nm. 2a showed a closely related profile to 1a, characterized by an intense band at 430 nm (ε = 3.57 × 104 L mol−1 cm−1) together with a broad absorption in the 450–520 nm region. To gain further insight into this feature, TD-DFT calculations were performed at the B3LYP/6-31+G(d,p)-CPCM(CH2Cl2) level of theory, employing model substrates 1a′ and 2a′ with tBu groups omitted (Fig. 6c). The calculated HOMO and LUMO of 1a′ were similar to those of typical dibenzo[j,l]fluoranthenes.38 The HOMO of 2a′ was localized on one fluoranthene unit, while the LUMO was delocalized over both units. The HOMO–LUMO energy gaps (ΔEHOMO–LUMO, eV) and the oscillator strengths (f) are in good agreement with observed trends in UV-vis absorption. In the fluorescence spectrum, 2a exhibited a slightly red-shifted emission maximum (λem = 584 nm) compared to 1a (λem = 575 nm). The fluorescence quantum yield (Φ) of 2a (2.6%) was higher than that of 1a (1.7%). Because both 1a and 2a are intrinsically rigid and undergo little structural change upon excitation, the enhanced fluorescence efficiency of 2a can be ascribed to its more allowed S0–S1 transition, as reflected by the larger oscillator strength obtained from TD-DFT calculations.
 |
| | Fig. 6 (a) CD spectra of 2a in CHCl3 at 298 K. (b) UV-vis absorption (solid line) and fluorescence (dashed line) spectra of 1a and 2a in CH2Cl2 at 298 K. (c) Calculated frontier molecular orbitals, energy levels (ΔEHOMO–LUMO) and oscillator strengths (f) of 1a′ and 2a′ (tBu groups are omitted) at the B3LYP/6-31+G (d,p)-CPCM(CH2Cl2) level of theory. The contribution of the HOMO to LUMO transition is shown in parentheses below the f value. | |
Conclusions
In summary, we have developed a Brønsted acid, light, and O2 driven protocol for oxidative C–H/C–H biaryl coupling from π-extended arenols. This method provides direct access to axially chiral 3D NGs, including those derived from non-benzenoid fluoranthene scaffolds. The scope of this reaction extends to a variety of arenols beyond fluoranthenes, underscoring the generality of this approach. Mechanistic studies revealed that the transformation proceeds via radical cation intermediates, while computational analysis predicted the formation of a self-induced pseudo-homo CT complex under acidic conditions, rationalizing the observed reactivity. This metal-free and direct C–H coupling provides a general strategy for designing π-extended chiral NGs with tailored photophysical and chiroptical properties.
Author contributions
K. K. discovered the reaction and performed the experiments, measurements and calculations. K. T. directed the project with A. K., Y. K. and H. T. K. T. -K. helped to perform ESR experiment and discussed. K. K. prepared the manuscript with input from all authors.
Conflicts of interest
There are no conflicts to declare.
Data availability
CCDC 2498496, 2498947, 2498498, 2498499, 2498500, 2498501, and 2498502 (1a, 1b, 1d, 2a, 2d, 2f and 4a) contain the supplementary crystallographic data for this paper.45a–g
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc09144f.
Acknowledgements
This study was supported by a JSPS KAKENHI (23H02604; KT), MEXT KAKENHI (21H05211; KT) in Digi-TOS, BINDS from AMED (22ama121034j0001; KT), and JST SPRING (JPMJSP2110; KK).
Notes and references
- V. Kumar, J. L. Páez, S. Míguez-Lago, J. M. Cuerva, C. M. Cruz and A. G. Campaña, Chem. Soc. Rev., 2025, 54, 4922–4947 RSC.
- C. Zhang, Y. Liu, X.-Q. Xiong, L.-H. Peng, L. Gan, C.-F. Chen and H.-B. Xu, Org. Lett., 2012, 14, 5912–5915 CrossRef CAS PubMed.
- B.-L. Hu, C. An, M. Wagner, G. Ivanova, A. Ivanova and M. Baumgarten, J. Am. Chem. Soc., 2019, 141, 5130–5134 CrossRef CAS PubMed.
- D. P. Sumy, N. J. Dodge, C. M. Harrison, A. D. Finke and A. C. Whalley, Chem. Eur J., 2016, 22, 4709–4712 CrossRef CAS PubMed.
- R. Kumar, P. J. Chmielewski, T. Lis, D. Volkmer and M. Stępień, Angew. Chem., Int. Ed., 2022, 61, e202207486 CrossRef CAS PubMed.
- J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernández, A. Molina-Ontoria, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2020, 142, 4162–4172 CrossRef CAS.
- P. Liu, H.-H. Lv, R. Xue, X.-Y. Tang, X.-Y. Chen, L. Qi and X.-Y. Wang, Org. Mater., 2024, 6, 40–44 CrossRef CAS.
- P. Izquierdo-García, J. M. Fernández-García, J. Perles, I. Fernández and N. Martín, Angew. Chem., Int. Ed., 2023, 62, e202215655 CrossRef PubMed.
- J. Lión-Villar, J. M. Fernández-García, S. M. Rivero, J. Perles, S. Wu, D. Aranda, J. Wu, S. Seki, J. Casado and N. Martín, Nat. Chem., 2025, 17, 1099–1106 CrossRef PubMed.
- K. K. Laali, T. Okazaki and S. E. Galembeck, J. Chem. Soc., Perkin Trans. 2, 2002, 621–629 RSC.
- P. Izquierdo-García, J. M. Fernández-García, I. Fernández, J. Perles and N. Martín, J. Am. Chem. Soc., 2021, 143, 11864–11870 CrossRef PubMed.
- X. Xu, S. Gunasekaran, S. Renken, L. Ripani, D. Schollmeyer, W. Kim, M. Marcaccio, A. Musser and A. Narita, Adv. Sci., 2022, 9, 2200004 CrossRef CAS PubMed.
- S. Li, R. Li, Y.-K. Zhang, S. Wang, B. Ma, B. Zhang and P. An, Chem. Sci., 2023, 14, 3286–3292 RSC.
- L. Qin, J. Xie, B. Wu, H. Hong, S. Yang, Z. Ma, C. Li, G. Zhang, X.-S. Zhang, K. Liu and D. Zhang, J. Am. Chem. Soc., 2024, 146, 12206–12214 CrossRef CAS PubMed.
- X. Zhang, H. Ma, Q. Yin, F. Bai, Y. Hashikawa and Chaolumen, Org. Lett., 2025, 27, 8969–8973 CrossRef CAS PubMed.
- K. Nakano, Y. Hidehira, K. Takahashi, T. Hiyama and K. Nozaki, Angew. Chem., Int. Ed., 2005, 44, 7136–7138 CrossRef CAS PubMed.
- M. Sako, Y. Takeuchi, T. Tsujihara, J. Kodera, T. Kawano, S. Takizawa and H. Sasai, J. Am. Chem. Soc., 2016, 138, 11481–11484 CrossRef CAS PubMed.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- J. Vercammen, M. Bocus, S. Neale, A. Bugaev, P. Tomkins, J. Hajek, S. V. Minnebruggen, A. Soldatov, A. Krajnc, G. Mali, V. V. Speybroeck and D. E. D. Vos, Nat. Catal., 2020, 3, 1002–1009 CrossRef CAS.
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163–169 CrossRef CAS.
- X. Wang, X. Xun, H. Song, Y. Liu and Q. Wang, Org. Lett., 2022, 24, 4580–4585 CrossRef CAS PubMed.
- T. Takada, M. Arisawa, M. Gyoten, R. Hamada, H. Tohma and Y. Kita, J. Org. Chem., 1998, 63, 7698–7706 CrossRef CAS.
- T. Dohi, K. Morimoto, Y. Kiyono, A. Maruyama, H. Tohma and Y. Kita, Chem. Commun., 2005, 2930–2932 RSC.
- A. Kumar, H. Sasai and S. Takizawa, Acc. Chem. Res., 2022, 55, 2949–2965 CrossRef CAS PubMed.
- J. Wu, Y. Koga, G. Hong, M. E. Rotella and M. C. Kozlowski, J. Am. Chem. Soc., 2025, 147, 34089–34100 CrossRef CAS PubMed.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–897 CrossRef CAS PubMed.
- J.-M. Tian, A.-F. Wang, J.-S. Yang, X.-J. Zhao, Y.-Q. Tu, S.-Y. Zhang and Z.-M. Chen, Angew. Chem., Int. Ed., 2019, 58, 11023–11027 CrossRef CAS PubMed.
- S.-H. Wang, S.-Q. Wei, Y. Zhang, X.-M. Zhang, S.-Y. Zhang, K.-L. Dai, Y.-Q. Tu, K. Lu and T.-M. Ding, Nat. Commun., 2024, 15, 4591 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, Z. Song, K. Nakajima and T. Takahashi, Chem. Lett., 2013, 42, 697–699 CrossRef CAS.
- S. Takizawa, J. Kodera, Y. Yoshida, M. Sako, S. Breukers, D. Enders and H. Sasai, Tetrahedron, 2014, 70, 1786–1793 CrossRef CAS.
- K. Hassan, K. Yamashita, K. Hirabayashi, T. Shimizu, K. Nakabayashi, Y. Imai, T. Matsumoto, A. Yamano and K. Sugiura, Chem. Lett., 2015, 44, 1607–1609 CrossRef CAS.
- M. R. Lasky, T. K. Salvador, S. Mukhopadhyay, M. S. Remy, T. P. Vaid and M. S. Sanford, Angew. Chem., Int. Ed., 2022, 61, e202208741 CrossRef CAS PubMed.
- D. S. Brandes, M. R. Lasky, A. V. R. D. Lisboa, E.-C. Liu, M. S. Remy and M. S. Sanford, Org. Lett., 2025, 27, 10011–10015 CrossRef CAS PubMed.
- M. C. Carson, C. R. Liu, Y. Liu and M. C. Kozlowski, ACS Catal., 2024, 14, 12173–12180 CrossRef CAS PubMed.
- M. C. Carson, C. R. Liu and M. C. Kozlowski, J. Org. Chem., 2024, 89, 3419–3429 CrossRef CAS PubMed.
- N. Ogawa, Y. Yamaoka, K. Yamada and K. Takasu, Org. Lett., 2017, 19, 3327–3330 CrossRef CAS PubMed.
- N. Ogawa, Y. Yamaoka, H. Takikawa, K. Yamada and K. Takasu, J. Am. Chem. Soc., 2020, 142, 13322–13327 CrossRef CAS PubMed.
- K. Kurokawa, N. Ogawa, Y. Kuroda, Y. Yamaoka, H. Takikawa, K. Tsubaki and K. Takasu, Org. Biomol. Chem., 2024, 22, 5306–5313 RSC.
- Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684–3691 CrossRef CAS.
- A. M. Genaev, L. N. Shchegoleva, G. E. Salnikov, A. V. Shernyukov, L. A. Shundrin, I. K. Shundrina, Z. Zhu and K. Y. Koltunov, J. Org. Chem., 2019, 84, 7238–7243 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- H. Tohma, M. Iwata, T. Maegawa and Y. Kita, Tetrahedron Lett., 2002, 43, 9241–9244 CrossRef CAS.
- H. Choi, S.-E. Suh and H. Kang, Adv. Synth. Catal., 2024, 366, 4347–4384 CrossRef CAS.
- L. Meca, D. Řeha and Z. Havlas, J. Org. Chem., 2003, 68, 5677–5680 CrossRef CAS PubMed.
-
(a)
CCDC 2498496: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwn3;
(b)
CCDC 2498497: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwp4;
(c)
CCDC 2498498: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwq5;
(d)
CCDC 2498499: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwr6;
(e)
CCDC 2498500: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvws7;
(f)
CCDC 2498501: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwt8;
(g)
CCDC 2498502: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pvwv9.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Aki
Kohyama
a,
Aki
Kohyama







