
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of electron transfer pathways in atomically precise metal nanoclusters: catalyzing a leap in solar water oxidation
Peng
Su
a,
Jia-Liang
Liu
a and
Fang-Xing
Xiao
 *ab
*ab
aCollege of Materials Science and Engineering, Fuzhou University, New Campus, Minhou, Fujian Province 350108, China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, PR China. E-mail: fxxiao@fzu.edu.cn
First published on 15th December 2025
Abstract
Atomically precise metal nanoclusters (NCs, <2 nm) with precise numbers of metal atoms and ligands have attracted enormous attention as highly promising photosensitizers owing to their peculiar atom-stacking mode, quantum confinement effect, enriched active sites and discrete molecular-like energy band structures. Despite these merits, metal NCs still suffer from inherent drawbacks such as ultrafast photogenerated carrier recombination, poor photostability and difficulties in regulating the charge transport pathway, which severely hinder their applications in photocatalysis. Herein, we strategically constructed spatially hierarchically ordered MOs/(PDDA/MQDs/PDDA/Agy)n heterostructure artificial photosystems by layer-by-layer assembly of oppositely charged poly(diallyldimethylammonium chloride) (PDDA), MXene quantum dot (MQD) and Agy NC [Agx@GSH, Ag9(GSH)6, and Ag16(GSH)9] building blocks on a metal oxide (MO) substrate under ambient conditions for customizing the directional charge migration/separation pathway over atomically precise metal NCs. In these on-demand artificial photosystems, Agy NCs serve as highly efficient photosensitizers that markedly enhance the visible light absorption capacity, while the synergistic and concurrent electron-withdrawing roles of PDDA (as an electron extraction medium) and MQDs (as electron acceptors) contribute to the long-range tandem charge transport chain, synergistically driving the directional electron transfer from the metal NCs to the MO framework. This significantly accelerates the charge separation/transfer of metal NCs and markedly improves the solar water oxidation performances of the MOs/(PDDA/MQDs/PDDA/Agy)n heterostructures. The essential roles of each building block are specifically explored with the photoelectrochemical mechanism clearly elucidated. Our work offers an accessible and universal route to strategically construct metal NC-based heterostructure photosystems and unveils the multi-functional synergy in regulating the electron migration pathway of atomically precise metal NCs towards solar energy conversion.
1 Introduction
Atomically precise metal nanoclusters (NCs, <2 nm) have been attracting enormous attention due to their unique atom-stacking fashion, quantum-confinement effect and abundant active sites.1–4 In particular, the discrete molecule-like energy band structure renders metal NCs ideal photosensitizers for driving photoredox reactions, markedly enhancing their light-harvesting ability and photocatalytic performances.5–8 Thus far, fine-tuning of photoinduced charge transfer of metal NCs in photoelectrochemical (PEC) water oxidation has been poorly explored. This is mainly due to the inherent limitations of metal NCs including ultrashort photogenerated carrier lifetime, poor stability and difficulty in regulating the charge transport pathway, substantially hindering the construction of high-performance metal NC-based artificial photosystems.9Applicable interface engineering is the key to access the delicate regulation of the charge transport pathway over atomically precise metal NCs. Coupling metal NCs with metal oxides (MOs) in fabricating well-defined hetero-interfaces with matched energy level alignment can effectively broaden the spectral response range of MOs and simultaneously enhance the charge separation in metal NCs.10,11 From the perspective of structural features of metal NCs, the thiolate ligands capped on the metal NC surface offer a flexible platform for their pinpoint self-assembly on the MO substrate via molecular interaction, enabling the easily accessible, facile, and efficient construction of an elegant interface between them. This calls for suitable materials-fabricating technology. Layer-by-Layer (LbL) assembly demonstrates unprecedented advantages in fabricating spatially multilayer heterostructures in terms of simplicity, versatility, and universality.12,13 More intriguingly, it is performed by judiciously harnessing the surface charge properties of building blocks via molecular interaction under merely ambient conditions, surpassing the conventional synthetic approaches involving tedious, complicated and harsh experimental conditions such as high temperature and pressure as well as calcination. Considering the necessity of customizing well-defined hetero-interfaces, stabilizing the structural integrity of metal NCs and crafting high-performance metal NC/MO PEC artificial photosystems, the LbL assembly strategy offers an ideal route for the controlled construction of metal NC/MO multilayer heterostructures.
Rational selection of applicable assembly units to integrate with metal NCs in LbL assembly of metal NC/MO heterostructures is of paramount importance for customizing a high-quality interface that enables fine-tuning of charge transport over metal NCs. Notably, polyelectrolytes (PEs) have been widely utilized in LbL assembly buildup as “molecular glue” to enable close integration of diverse building blocks, but their roles as interfacial charge transport mediators have been underestimated for a long time.14,15 Our group has ascertained that certain PEs can function as electron-withdrawing media to accelerate charge separation in semiconductors.3,16 As such, PEs as building blocks introduced in LbL-assembled artificial photosystems could act as robust co-catalysts to benefit the interfacial charge transfer of metal NCs. On the other hand, in recent years, MXene quantum dots (MQDs) have gained enormous attention for possessing abundant edge active sites, diverse surface states and excellent conductivity, which demonstrate their great potential as efficient electron acceptors.17,18 As a result, the generic merits of PEs and MQDs make them desirable electron migration regulators to facilitate charge transportation and separation in metal NCs.19–23 Based on the above analysis, we speculate that if PEs, MQDs, and atomically precise metal NCs are selected as building blocks and MOs as substrates onto which electron-trapping PEs and MQDs are concurrently integrated with metal NCs via LbL assembly, the electron migration pathway of metal NCs can be synergistically and precisely tuned by the stacking mode and assembly sequence of PEs and MQDs, thereby driving long-range tandem electron transfer from the metal NCs to the MO substrate, markedly enhancing the charge separation in metal NCs, and ultimately boosting the PEC water splitting performances.
Herein, with these considerations in mind, we develop a facile and easily accessible LbL assembly strategy to construct TiO2/(PDDA/MQDs/PDDA/Agy)n multilayer heterostructure photoanodes under merely ambient conditions. In these well-designed heterostructures, oppositely charged poly(dimethylamino)propyl chloride ammonium (PDDA), Agy NC and MQD building blocks are deposited in a highly ordered manner on the MO substrate to construct a spatially ordered cascade charge transport chain. Among them, Agy NCs [Agx@GSH, Ag9(GSH)6, and Ag16(GSH)9] act as highly efficient photosensitizers for visible light harvesting and charge carrier generation. Simultaneously, PDDA and MQDs concurrently serve as the synergistic electron-withdrawing mediators to strengthen the electron transport impetus from Agy NCs to the TiO2 substrate, hence accelerating the charge transport and separation over Agy NCs. Our work provides inspiring ideas for the rational construction of a robust and efficient metal NC-based artificial PEC system and for mediating tunable charge separation over atomically precise metal NCs towards solar energy conversion.
2 Experimental section
2.1 Preparation of the TNTA substrate2
TNRA substrates were prepared by a two-step electrochemical anodization method. The detailed preparation process is provided in the SI.2.2 Preparation of metal NCs24
Detailed information on the preparation of Agx@GSH NCs, Ag9(GSH)6 NCs and Ag16(GSH)9 NCs is provided in the SI.2.3 Preparation of Ti3C2 MQDs16
2 g of Ti3AlC2 powder was immersed in 40 mL of 48% HF and stirred for 20 h at 60 °C. The obtained powder was then rinsed multiple times with deionized water (DI H2O). The powder pellet was retrieved by centrifugation at 3500 rpm for 10 min, and the supernatant was discarded. The final product (Ti3C2 MXene powder) was dried at 60 °C under vacuum for 12 h. In an N2-protected environment, the Ti3C2 MXene powder was then placed in 80 mL DI H2O, followed by ultrasound treatment for 1 h. The pH of the mixture was adjusted to around 9 using ammonium hydroxide and the mixture was transferred into a 50 mL Teflon-lined autoclave at 120 °C for 6 h. After that, the MQDs were obtained by filtering the mixture through a 220 nm membrane.2.4 Preparation of TNTA/(P/MQDs/P/Agy)n photoanodes
The fabrication of multilayer composite photoelectrodes is achieved by an electrostatic self-assembly approach under ambient conditions. First, TiO2 nanotube array (TNTA) substrates were dipped into a PDDA aqueous solution (0.5 mg mL−1, 0.5 M NaCl, pH = 10) for 30 s, and washed with DI H2O for 1 min to obtain TNTA/(P)1. Then, TNTA/(P)1 was immersed in a Ti3C2 MQD aqueous solution (24.6 mg mL−1, pH = 7) for 5 min, and washed with DI H2O for 1 min to obtain TNTA/(P/MQDs)1. Then, TNTA/(P/MQDs)1 was immersed in PDDA aqueous solution for another 30 s, and washed with DI H2O for 1 min leading to TNTA/(P/MQDs P−1)1. It was re-immersed into a Agx@GSH NCs aqueous solution (0.12 mg mL−1) for 5 min and washed with DI H2O. The above process was defined as one assembly cycle, i.e., TNTA/(P/MQDs/P/Agx)1. The above procedures were repeated to obtain the TNTA/(P/MQDs/P/Agx)n multilayer heterostructure photoanodes with a designated assembly cycle. The counterpart of TNRAs/(Agx)n photoanodes was also prepared by loading Agx@GSH NCs on the TNTA substrate. Under the same conditions, Agx@GSH NCs were replaced with Ag9(GSH)6 NCs and Ag16(GSH)9 NCs to construct the TNTA/(P/MQDs/P/Agy)n [Agy = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9] multilayer heterostructures.2.5 Characterization
The crystal phase was determined by X-ray diffraction (XRD, Miniflex600). Scanning electron microscopy (SEM, Supra55) was applied to probe the morphological features of the photoelectrodes. Transmission electron microscopy (TEM) images were obtained using a transmission electron microscope (Tecnai G2 F20). Optical properties of the photoelectrodes were analyzed by studying UV-vis diffuse reflectance spectra (Cary7000 UV-visible spectrophotometer, Varian, America), which were obtained utilizing BaSO4 as the reflectance background ranging from 200 to 800 nm. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a photoelectron spectrometer (ESCALAB 250, Thermo Scientific, America), where binding energy (BE) of the elements was calibrated by the BE of C 1s (284.80 eV). Raman spectra were recorded using a Raman spectrometer (Dxr-2xi). Fourier transform infrared (FTIR) spectra were recorded using a TJ270-30A infrared spectrophotometer (Tianjin, China). Photoluminescence spectra (PL) for solid samples were obtained using an Edinburgh photoluminescence spectrophotometer. Time-resolved photoluminescence (TRPL) spectra were recorded using an FS5 fluorescence lifetime spectrophotometer (Edinburgh Instruments, UK).2.6 PEC water oxidation measurements
PEC water splitting measurements were performed using a conventional three-electrode configuration on an electrochemical workstation (Interface 1000E, Gamry, America). TNTA and TNTA/Agy NCs (Agy = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9) (1 cm × 1 cm) were directly employed as the working electrode with Pt foil and a Ag/AgCl electrode as the counter and reference electrodes, respectively. The electrolyte is composed of a Na2SO4 (0.5 M, pH = 6.69) aqueous solution. PEC water splitting performances were evaluated under simulated sunlight irradiation from a 300 W Xe lamp (PLS FX300HU, Beijing Perfect Light co. Ltd, China) equipped with a UV-cutoff filter for visible light (λ > 420 nm), which was employed as the light source for visible light irradiation. The potentials of the electrodes are corrected against the reversible hydrogen electrode (RHE) via the calibration equation as shown below:| ERHE = EAg/AgCl + 0.059 pH + E°Ag/AgCl | (1) |
| (E°Ag/AgCl = 0.1976 V at 25 °C) |
3 Results and discussion
3.1 Characterization of composite photoanodes
The synthetic process for the fabrication of TNTA/(PDDA/MQDs/PDDA/Agy)n [Agy = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9] multilayer heterostructure photoanodes is illustrated in Fig. 1a and S1–S3. First, an ultrathin PDDA layer (Fig. S4) was deposited onto the TNTA substrate via molecular interaction to form the TNTA/(PDDA), imparting a uniform positively charged surface. Subsequently, negatively charged MQDs (Fig. S5) were deposited on the surface of TNTA/(PDDA) through electrostatic attraction to form the TNTA/(PDDA/MQDs) heterostructure. Next, the surface of TNTA/(PDDA/MQDs) was further modified using PDDA to re-attain the positively charged surface, thereby facilitating the electrostatic attraction of negatively charged Agy@GSH NCs [Agy = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9]. It is noteworthy that the negative charge on the surface of Agy@GSH NCs originates from the deprotonation of the GSH ligands (Fig. S6). Therefore, the negatively charged Agy@GSH NCs can serve as an assembly module and effectively combine with the oppositely charged TNTA/(PDDA/MQDs/PDDA), ultimately giving rise to the spatially multilayered TNTA/(PDDA/MQDs/PDDA/Agy)1 heterostructures. Furthermore, by repeating the PDDA-MQDs-PDDA-Agy@GSH NCs assembly cycle mentioned before, TNTA/(PDDA/MQDs/PDDA/Agy)n heterostructure photoanodes were constructed. In this multilayered heterostructure, the loading amount of MQDs and Agy@GSH NCs can be precisely controlled by adjusting the number of assembly cycles. | ||
| Fig. 1 (a) Schematic demonstration of LbL assembly of TNTA/(P/MQDs/P/Agy)n (y = x, 9, 16) multilayer heterostructures. SEM images of (b) TNTA, (c) TNTA/(Agx)6 and (d) TNTA/(P/MQDs/P/Agx)6 with corresponding cross-sectional images. (e) TEM and (f & g) HRTEM images of TNTA/(P/MQDs/P/Agx)6 with (h–o) corresponding mapping results. | ||
The morphologies of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)n were analyzed by scanning electron microscopy (SEM). Fig. 1b presents the top-view SEM image of TNTA, which exhibits a uniform and regular honeycomb-like structure composed of highly ordered nanotubes. The cross-sectional view (inset) clearly reveals that the nanotube array possesses a vertical height of approximately 5.5 µm. Fig. 1c shows the top-view SEM image of TNTA/(Agx)6. The morphology of TNTA/(Agx)6 remains nearly identical to that of pristine TNTA, which is attributed to the ultra-small size of Agx@GSH NCs (ca. 2.4 nm) surpassing the resolution limit of SEM measurement (Fig. S7). However, the SEM elemental mapping pattern of TNTA/(Agx)6 (Fig. S8) shows that in addition to the Ti and O elements from the TNTA substrate, Ag, S, and N elements were also detected, confirming the successful loading of Agx@GSH NCs on the TNTA substrate. It should be emphasized that the size of MQDs is also small (ca. 4 nm, Fig. S9) making direct differentiation of it from Agx@GSH NCs very difficult. Notably, the top-view SEM image of TNTA/(P/MQDs/P/Agx)6 (Fig. 1d) differs from those of TNTA and TNTA/(Agx)6, revealing subtle protrusions inside the nanotubes. This morphological feature arises from the multilayer assembly of PDDA, Agx@GSH NC and MQD building blocks, providing direct evidence that PDDA, MQD and Agx@GSH NC building blocks have been successfully deposited onto the TNTA substrate (Fig. 1e). Consistently, characteristic elemental signals including Cl from PDDA, Ag from Agx@GSH NCs and F from MQDs are detected in the SEM elemental mapping pattern of TNTA/(P/MQDs/P/Agx)6 (Fig. S10). Transmission electron microscopy (TEM) was further employed to verify the assembly of PDDA, MQDs and Agx@GSH NCs on the TNTA substrate. As shown in Fig. 1f, MQDs and Agx@GSH NCs were successfully loaded onto the TNTA surface, and an ultra-thin PDDA layer with thickness of approximately 4 nm is observed. Fig. 1g displays the HRTEM image of TNTA/(P/MQDs/P/Agx)6 wherein the lattice fringes of 0.352 nm and 0.247 nm correspond to the (101) crystal plane of TiO2 and (006) plane of MQDs, respectively.25 However, no lattice fringes attributable to Agx@GSH NCs are observed, consistent with their distinctive atom-packing mode. TEM elemental mapping was employed to investigate the distribution of elements in TNTA/(P/MQDs/P/Agx)6. As shown in Fig. 1h–o, Ti, O, S, Cl, Ag, F and N are uniformly distributed on the TNTA substrate, among which Ti & O arise from the TNTA substrate, the Ag signal is from the Agx@GSH NCs, Cl originates from the PDDA, N & S are shared by both the PDDA and GSH ligands of Agx@GSH NCs, while F originates from MQDs. These results provide compelling evidence that PDDA, MQD and Agx@GSH NC building blocks have been successfully self-assembled on the TNTA substrate, thereby forming the TNTA/(P/MQDs/P/Agx)6 multilayer heterostructure photoanode.
To elucidate the crystal structures of the samples, TNTA, TNTA/(Agx)6, and TNTA/(P/MQDs/P/Agx)6 were systematically characterized by X-ray diffraction (XRD) (Fig. S11). XRD patterns of the TNTA substrate show peaks at 25.31°, 37.81°, 48.07°, 53.91° and 70.57°, all matching the anatase TiO2 (JCPDS 21-1272).26 XRD patterns of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 are analogous to that of pristine TNTA, with neither extra diffraction peaks nor peak shifts. This indicates that the loading of PDDA, MQDs and Agx@GSH NCs does not affect the crystal structure of the TNTA substrate, which might be due to the relatively low content of these assembly units. Raman spectra in Fig. 2a exhibit peaks at 144.5, 399, 518.1, and 639.4 cm−1, which correspond to the Eg, B1g, A1g + B1g, and Eg vibrational modes of anatase TiO2, respectively.27 Similarly, no characteristic peaks of PDDA, MQDs and Agx@GSH NCs are observed in the Raman spectra, further confirming the relatively low deposition amount of PDDA, MQDs and Agx@GSH NCs on the TNTA substrate. Notably, peak intensity of the samples shows a gradient attenuation trend of TNTA > TNTA/(Agx)6 > TNTA/(P/MQDs/P/Agx)6. This strongly indicates that deposition of PDDA, MQDs and Agx@GSH NCs exerts a shielding effect on the TNTA substrate, resulting in a decrease in signal intensity. As shown in Fig. 2b, the FTIR spectrum of the TNTA substrate displays three peaks at 1110, 1635, and 3430 cm−1, corresponding to the asymmetric stretching mode of the Ti–O bond from the TNTA substrate and the stretching & bending modes of surface hydroxyl (–OH) functional groups, respectively.13,16,28 Compared with TNTA, the diminished Ti–O signal in the FTIR spectrum of TNTA/(Agx)6 is attributed to the shielding effect of the deposited Agx@GSH NCs. Meanwhile, a new spectral band appears at 1407 cm−1, which corresponds to the symmetric stretching vibration mode of the –COOH group in the GSH ligands, confirming the successful anchoring of Agx@GSH NCs on the TNTA substrate.29 Further comparison reveals that TNTA/(P/MQDs/P/Agx)6 exhibits markedly intensified peaks at 1110 and 1635 cm−1 relative to TNTA/(Agx)6. Specifically, enhancement of the peak at 1110 cm−1 is related to the stretching vibration of the C–F bond in MQDs,16,30 while the significant increase in the peak intensity at 1635 cm−1 is attributed to the overlap of –NH2 bending vibration in Agx@GSH NCs with the intrinsic –OH band.3,24,31 The above results verify that Agx@GSH NCs and MQDs were successfully assembled onto the surface of the TNTA substrate through a LbL assembly strategy, thereby giving rise to the TNTA/(P/MQDs/P/Agx)6 heterostructure. UV-visible diffuse reflectance spectra (DRS) of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 are shown in Fig. 2c, in which the wide-band-gap TNTA substrate exhibits limited light absorption within the UV light scope. In contrast, both TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 exhibit markedly enhanced visible light absorption, which is mainly attributed to the photosensitization effect of Agx@GSH NCs (Fig. S12a). It is worth noting that the DRS spectra of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 are very similar. This indicates that MQD and PDDA decoration does not alter the overall light absorption capacity of the TNTA/(P/MQDs/P/Agx)6 heterostructure. As displayed in Fig. S12b, PDDA, as a polymer, exhibits no optical response within the visible range. In addition, MQDs also show negligible absorption in this region (Fig. S12c). These results support the photosensitization effect of Agx@GSH NCs, which plays a pivotal role in extending the photoresponse range and enhancing the visible-light-driven PEC performances of heterostructure photoanodes, which will be elucidated in the latter part.
 | ||
| Fig. 2 (a) Raman, (b) FTIR and (c) DRS results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6; high-resolution (d) Ti 2p, (e) O 1s, (f) Ag 3d, (g) C 1s of (II) TNTA/(Agx)6 and (III) TNTA/(P/MQDs/P/Agx)6, and high-resolution (h) S 2p, (i) N 1s, (j) Cl 2p and (k) F 1s of TNTA/(P/MQDs/P/Agx)6. | ||
X-ray photoelectron spectroscopy (XPS) was used to determine the composition and elemental chemical states of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6. As shown in Fig. S13, the survey spectrum of TNTA/(P/MQDs/P/Agx)6 exhibits the signals of F, Cl, N, S, Ag, C, Ti and O, among which the element Ag originates from Agx@GSH NCs, Cl is from the ultrathin PDDA layer, and F comes from MQDs, confirming the successful deposition of PDDA, MQDs and Agx@GSH NCs onto the TNTA substrate. Fig. 2d and e present the high-resolution Ti 2p and O 1s spectra of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6, respectively. To be specific, the binding energies at 458.2 eV (Ti 2p3/2) and 463.8 eV (Ti 2p1/2) for TNTA/(Agx)6 confirm the presence of Ti4+ species,32 while the O 1s spectrum shows two distinct peaks at 529.28 and 531.02 eV, attributed to the lattice oxygen (Ti–O) and surface hydroxyl (Ti–OH), respectively.27 High-resolution Ag 3d spectra of TNTA/(Agx)6 (Fig. 2f) show the peaks at 366.9 and 372.9 eV attributed to Ag+ species, while those at 367.7 and 373.7 eV correspond to Ag0.24 Notably, compared with TNTA/(Agx)6, Ti 2p, O 1s and Ag 3d spectra of TNTA/(P/MQDs/P/Agx)6 exhibit slight negative shifts in binding energy, indicative of the electronic interactions among PDDA, Agx@GSH NCs, MQDs and the TNTA substrate during the LbL assembly of the TNTA/(P/MQDs/P/Agx)6 multilayer heterostructure. Furthermore, high-resolution C 1s spectra of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 (Fig. 2g) exhibit the peaks assignable to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–F bonds, providing direct evidence for the presence of GSH ligands and MQDs.8,13,16 In the high-resolution S 2p spectrum of TNTA/(P/MQDs/P/Agx)6 (Fig. 2h), the formation of Ag–S bonds can be observed to confirm the successful deposition of Agx@GSH NCs on the TNTA substrate.33Fig. 2i presents the high-resolution N 1s spectrum of TNTA/(P/MQDs/P/Agx)6, with peaks positioning at 399.4 eV and 401.85 eV, corresponding to the –NH2/–NH– and –NH4+– species from the PDDA layer, respectively.3,34 Meanwhile, the high-resolution Cl 2p spectrum of TNTA/(P/MQDs/P/Agx)6 (Fig. 2j) shows the peaks at 196.7 eV and 198.2 eV that arise from the residual chloride species within the PDDA.35 The high-resolution F 1s spectrum of TNTA/(P/MQDs/P/Agx)6 at 683.5 eV (Fig. 2k) corresponds to the C–Ti–F species from Ti3C2 MQDs.16,36 Consequently, XPS results (Table S2) unequivocally confirm that PDDA, MQDs and Agx@GSH NCs have been successfully deposited on the TNTA substrate via the LbL strategy leading to the TNTA/(P/MQDs/P/Agx)n heterostructure.
O and C–F bonds, providing direct evidence for the presence of GSH ligands and MQDs.8,13,16 In the high-resolution S 2p spectrum of TNTA/(P/MQDs/P/Agx)6 (Fig. 2h), the formation of Ag–S bonds can be observed to confirm the successful deposition of Agx@GSH NCs on the TNTA substrate.33Fig. 2i presents the high-resolution N 1s spectrum of TNTA/(P/MQDs/P/Agx)6, with peaks positioning at 399.4 eV and 401.85 eV, corresponding to the –NH2/–NH– and –NH4+– species from the PDDA layer, respectively.3,34 Meanwhile, the high-resolution Cl 2p spectrum of TNTA/(P/MQDs/P/Agx)6 (Fig. 2j) shows the peaks at 196.7 eV and 198.2 eV that arise from the residual chloride species within the PDDA.35 The high-resolution F 1s spectrum of TNTA/(P/MQDs/P/Agx)6 at 683.5 eV (Fig. 2k) corresponds to the C–Ti–F species from Ti3C2 MQDs.16,36 Consequently, XPS results (Table S2) unequivocally confirm that PDDA, MQDs and Agx@GSH NCs have been successfully deposited on the TNTA substrate via the LbL strategy leading to the TNTA/(P/MQDs/P/Agx)n heterostructure.
3.2 PEC water oxidation performances
To clarify how the synergistic effect among PDDA, MQDs and Agx@GSH NCs influences carrier separation, PEC water oxidation performances of TNTA/(P/MQDs/P/Agx)n (n = 2, 4, 6, 8) photoanodes with different assembly numbers were systematically investigated under visible light irradiation (λ > 420 nm). Through the linear sweep voltammetry (LSV) test (Fig. S14a), the dependence of photocurrents of TNTA/(P/MQDs/P/Agx)n on the assembly number was systematically probed. Among the TNTA/(P/MQDs/P/Agx)n photoanodes (n = 2, 4, 6, 8), TNTA/(P/MQDs/P/Agx)6 exhibits the optimal photocurrent. This result suggests that the synergistic effect among PDDA, MQDs and Agx@GSH NCs markedly accelerates the carrier separation, thereby substantially enhancing the PEC performances of the TNTA/(P/MQDs/P/Agx)n photoanodes. However, further increase in the assembly layer (n = 8) leads to an excessively thick PDDA coating and over-deposition of MQDs and Agx@GSH NCs, which increases the density of charge recombination sites, thereby suppressing the charge transfer efficiency. The transient on–off photocurrent (I–t) results (Fig. S14b) closely mirror the LSV photocurrent trend, corroborating the above conclusions. Based on the above systematic analysis, TNTA/(P/MQDs/P/Agx)6 is identified as the optimal photoanode and selected for further investigation.Fig. 3a presents the LSV curves of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6. Under visible-light irradiation (λ > 420 nm), the TNTA substrate shows a negligible photocurrent, indicating that it is scarcely photoexcited for carrier generation because of the large bandgap. In sharp contrast, TNTA/(Agx)6 exhibits a considerably enhanced photocurrent density, unequivocally verifying the photosensitization effect of Agx@GSH NCs in enhancing the light-harvesting capacity of TNTA. Notably, TNTA/(P/MQDs/P/Agx)6 exhibits the most pronounced photocurrent response, attributable to the synergistic regulation of directional carrier transport by PDDA and MQDs. Specifically, the ultrathin PDDA layer and MQDs function as efficient electron-withdrawing mediators, thereby driving the directional electron migration from Agx@GSH NCs to TNTA and markedly suppressing carrier recombination. The applied-bias photon-to-current efficiency (ABPE, η) of the photoanode was calculated from the LSV data using the following formula (formula (2)).37
| ABPE = Ip × (1.23 − |V|)/Plight | (2) |
 | ||
| Fig. 3 (a) LSV, (b) ABPE, (c) photocurrent (I–t), and (d) OCVD curves, (e) electron lifetime, (f) IPCE and (g) EIS results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6; (h) anodic transient dynamics of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6; (i) M–S results and (j) carrier densities (ND) of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6; (k) charge injection and (l) separation efficiency of TNTA and TNTA/(P/MQDs/P/Agx)6 under visible light irradiation (λ > 420 nm) (electrolyte: 0.5 M Na2SO4, pH = 6.69). | ||
Charge recombination kinetics of the photoanodes were explored by open-circuit voltage decay (OCVD) measurements. Notably, the decay rate of the OCVD curve directly reflects the carrier separation efficiency, with a faster decay rate indicating higher charge separation efficiency.12,38 As shown in Fig. 3d, TNTA/(P/MQDs/P/Agx)6 exhibits the optimal OCVD kinetics, indicating that the synergistic effect among the PDDA charge-withdrawing interlayer, MQD electron acceptor and Agx@GSH NC photosensitizer achieves highly efficient carrier separation. The quantitative relationship between the open-circuit voltage (VOC) and electron lifetime (τn) is calculated using the following equation (eqn (3)).12
 | (3) |
To evaluate the photon-to-electron conversion performance of photoanodes, incident photon-to-current efficiency (IPCE) was measured under monochromatic light irradiation. IPCE is defined by the following equation (eqn (4)):39
 | (4) |
The interfacial charge transport dynamics of the photoanodes were additionally assessed by electrochemical impedance spectroscopy (EIS) measurements. The radius of the semicircle in the EIS plot is closely related to the interfacial charge transfer resistance, that is, the smaller the radius, the lower the charge transfer resistance, and thus the higher interfacial charge separation efficiency. As shown in Fig. 3g, TNTA/(P/MQDs/P/Agx)6 exhibits the smallest semicircle radius compared to TNTA/(Agx)6 and pristine TNTA, indicating that the multilayered and spatially orderly interface structure constructed by alternately depositing PDDA, MQDs, and Agx NCs on the TNTA substrate fosters interfacial charge transfer at the heterostructure photoanode.40 According to the EIS fitting results (Table S3), the charge transfer resistance (Rct) of TNTA/(P/MQDs/P/Agx)6 is 1908 ohms, which is significantly lower than those of TNTA (9456 ohms) and TNTA/(Agx)6 (6070 ohms). This further strongly confirms that the multilayer heterostructure of TNTA/(P/MQDs/P/Agx)6 is conducive to promoting rapid interfacial charge transfer. Moreover, based on the I–t results (Fig. S17), a normalized parameter (D) was introduced to investigate the recombination behavior of charge carriers on the photoelectrode.41
 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D is equal to −1 (Fig. 3h). The τ value of TNTA/(P/MQDs/P/Agx)6 (4.71 s) is larger than that of TNTA/(Agx)6 (4.26 s), signifying that the cascade electron transfer pathway constructed by alternately depositing PDDA, MQDs and Agx@GSH NCs on the TNTA is conducive to extending the electron lifetime. Fig. 3i shows the Mott–Schottky (M–S) results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6. The positive slope of the M–S curves for all the samples indicates that they are typical n-type semiconductors.38 It has been widely accepted that the smaller the slope of the M–S plot, the larger the charge carrier density.3,11,13 Compared with TNTA and TNTA/(Agx)6, TNTA/(P/MQDs/P/Agx)6 exhibits the smallest M–S slope, indicating its largest carrier density. To quantify the carrier density (ND) of the photoelectrodes, the following formula was utilized:42
D is equal to −1 (Fig. 3h). The τ value of TNTA/(P/MQDs/P/Agx)6 (4.71 s) is larger than that of TNTA/(Agx)6 (4.26 s), signifying that the cascade electron transfer pathway constructed by alternately depositing PDDA, MQDs and Agx@GSH NCs on the TNTA is conducive to extending the electron lifetime. Fig. 3i shows the Mott–Schottky (M–S) results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6. The positive slope of the M–S curves for all the samples indicates that they are typical n-type semiconductors.38 It has been widely accepted that the smaller the slope of the M–S plot, the larger the charge carrier density.3,11,13 Compared with TNTA and TNTA/(Agx)6, TNTA/(P/MQDs/P/Agx)6 exhibits the smallest M–S slope, indicating its largest carrier density. To quantify the carrier density (ND) of the photoelectrodes, the following formula was utilized:42 | (6) |
The photocurrent  of PEC water splitting reaction can be expressed by eqn (7).3
of PEC water splitting reaction can be expressed by eqn (7).3
 | (7) |
where Jabs denotes the maximum photocurrent density achievable under 1 sun irradiation and internal quantum efficiency is 100% (Jabs of rutile TiO2 = 1.8 mA cm−2). ηinj and ηsep are defined as the hole injection efficiency for the target electrochemical reaction and the charge separation efficiency for transporting holes to the reaction surface, respectively. Using Na2SO3 as a hole scavenger, the ηsep and ηinj values of TNTA and TNTA/(P/MQDs/P/Agx)6 photoanodes were obtained using the following equations.3
 | (8) |
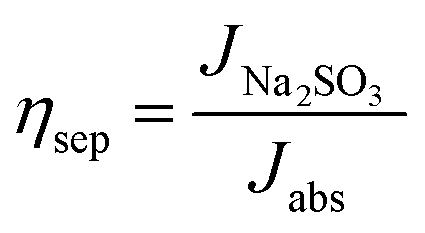 | (9) |
As shown in Fig. S18a–c, cyclic voltammetry (CV) was employed to systematically investigate the current responses of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 photoelectrodes at different scan rates (20, 40, 60, 80, and 100 mV s−1) within the non-faradaic potential range to evaluate their effective electrochemical active surface area (ECSA). The results indicate that all the photoelectrodes exhibit excellent symmetric capacitive properties, with their electrochemical response intensities increasing linearly with the scan rate. Based on the current density data at different scan rates (Fig. S18d–f), the corresponding double-layer capacitance values (Cdl) were obtained through linear fitting: 100.57 µF cm−2 for the TNTA substrate, 104.34 µF cm−2 for the TNTA/(Agx)6, and 110.59 µF cm−2 for the TNTA/(P/MQDs/P/Agx)6. Compared with the TNTA substrate, Cdl of TNTA/(Agx)6 is enhanced, indicating that Agx@GSH NC decoration increases the charge storage capacity at the interface and promotes the accumulation and transport of photogenerated electrons. Note that after introducing the PDDA and MQD building blocks to construct the TNTA/(P/MQDs/P/Agx)6 spatially multilayer heterostructure photoelectrode, the Cdl achieves an improvement (110.59 µF cm−2), which strongly confirms that LbL assembly of PDDA, MQDs and Agx@GSH NCs substantially enriches the interface active sites and optimizes the overall charge storage and transport of the photoelectrode.
Fig. 4a and b illustrate the stability of the TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 photoanodes under visible light irradiation. The results indicate that under continuous light irradiation for 1 h, photocurrents of these two photoanodes exhibit a decreasing trend. The decrease in the photocurrent of TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 photoanodes is related to the stability of Agx@GSH NCs, and it is possible that some Agx@GSH NCs may be destroyed to form Ag nanoparticles (NPs). Although both photoanodes exhibit a gradual decline in photocurrent, the alternating assembly structure provided by the LbL self-assembly design can offer a protective barrier for the Agx@GSH NC active layer, thereby inhibiting the oxidation corrosion it suffers during the PEC reaction. Consequently, TNTA/(P/MQDs/P/Agx)6 maintains a higher photocurrent (87.2 µA cm−2) and prolonged stability (2400 s). This strongly substantiates the significant superiority of PDDA and MQDs in enhancing the stability of composite photoelectrodes. In general, compared with TNTA/(Agx)6, the overall stability of the TNTA/(P/MQDs/P/Agx)6 photoanode is effectively improved. Furthermore, Table S4 demonstrates that the TNTA/(P/MQDs/P/Agx)6 photoanode, fabricated via a LbL self-assembly strategy, exhibits superior photoelectrochemical performance to previously reported TiO2-based photoanodes.
 | ||
| Fig. 4 Stability of (a) TNTA/(Agx)6 and (b) TNTA/(P/MQDs/P/Agx)6 under continuous visible light irradiation (λ > 420 nm) for 1 h; (c & d) LSV and (e & f) I–t results of TNTA and TNTA/(P/MQDs/P/Agy)6 (y = 9, 16) under visible light irradiation (λ > 420 nm); (g) schematic illustration of the fabrication of TNTA/(P/MQDs/P/Ag9)6 and TNTA/(P/MQDs/P/Ag16)6 heterostructure photoanodes. | ||
To assess the universality of the synergistic effect between metal NCs and electron-withdrawing mediators of PDDA/MQDs, Ag9(GSH)6 NCs and Ag16(GSH)9 NCs were also selected as the alternative photosensitizers for investigation (Fig. S19). Using the same LbL assembly strategy, similar multilayer heterostructures of TNTA/(P/MQDs/P/Ag9)6 and TNTA/(P/MQDs/P/Ag16)6 were successfully constructed (Fig. 4g). The morphologies (Fig. S20) of both TNTA/(P/MQDs/P/Ag9)6 and TNTA/(P/MQDs/P/Ag16)6 counterparts are analogous to that of TNTA/(P/MQDs/P/Agx)6. In addition, PEC water oxidation performances of TNTA/(P/MQDs/P/Ag9)6 and TNTA/(P/MQDs/P/Ag16)6 were probed under identical conditions. Under visible light irradiation, compared with the pristine TNTA substrate, PEC water oxidation activities of TNTA/(P/MQDs/P/Ag9)6 and TNTA/(P/MQDs/P/Ag16)6 are substantially enhanced (Fig. 4c–f). The results indicate that the synergistic effect of metal NCs [Agy NCs = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9] with PDDA & MQDs on enhancing the PEC water oxidation activity is universal.
PEC water oxidation activities of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 under simulated sunlight (AM1.5G) irradiation, in addition to visible light, were also systematically investigated. LSV results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 are shown in Fig. S21a, in which TNTA/(P/MQDs/P/Agx)6 still exhibits the optimal photocurrent response (i.e., TNTA/(P/MQDs/P/Agx)6 > TNTA/(Agx)6 > TNTA), consistent with the results probed under visible light irradiation. Besides, TNTA/(P/MQDs/P/Agx)6 also demonstrates the highest conversion efficiency (Fig. S21b), confirming that the synergistic effect among PDDA, MQDs and Agx@GSH NCs effectively enhances the separation and utilization efficiency of photogenerated charge carriers. In addition, I–t (Fig. S21c), EIS (Fig. S21d), OCVD (Fig. S21e) and electron lifetime (Fig. S21f) results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6 all show similar trends to the photocurrent. In summary, under simulated sunlight (AM1.5G) irradiation, TNTA/(P/MQDs/P/Agx)6 consistently exhibits the best PEC water splitting activities, which is consistent with the results obtained under visible light.
3.3 PEC water oxidation mechanism
The charge transport mechanism of the TNTA/(P/MQDs/P/Agy)6 multilayer heterostructure was further analyzed through in situ XPS testing (Fig. S22). Specifically, the shift of binding energy of the key elements towards a higher value indicates that the atoms in the photocatalyst have lost electrons, while a shift towards a lower value indicates that the atoms have gained electrons.44 As shown in Fig. 5a, under in situ visible light irradiation, binding energies of Ag 3d3/2 and Ag 3d5/2 shift towards higher values by 0.25 eV and 0.27 eV, respectively. This strongly confirms that Agx@GSH NCs, as efficient photosensitizers, provide photogenerated electrons in the TNTA/(P/MQDs/P/Agx)6 multilayer heterostructure to participate in the PEC water oxidation reaction. Meanwhile, the evolution of the N 1s binding energy precisely verifies the pivotal role of PDDA in stimulating the generation of the electron transfer chain in the ternary heterostructure. It is noteworthy that the binding energy of N 1s decreases by 0.31 eV and 0.16 eV under in situ light irradiation (Fig. 5b). This strongly corroborates its characteristics as an efficient electron transfer medium.3 Similarly, the binding energy of F 1s in MQDs also shows an apparent negative shift, additionally confirming their crucial role as an electron acceptor in regulating electron transport (Fig. 5c). The above results concurrently indicate that the cascade electron transport channel arising from the synergistic effect of PDDA and MQDs accelerates the directional electron migration from Agx@GSH NCs to TNTA, promoting the spatial separation of photogenerated charge carriers and enhancing the PEC water oxidation activity. | ||
| Fig. 5 High-resolution in situ XPS spectra of (a) Ag 3d, (b) N 1s and (c) F 1s for TNTA/(P/MQDs/P/Agx)6; (d) PL and (e) TRPL results of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6; (f) schematic illustration of the PEC water oxidation mechanism of TNTA/(P/MQDs/P/Agy)n photoanodes. | ||
Fig. 5d and e show the photoluminescence (PL) and time-resolved photoluminescence (TRPL) spectra of TNTA, TNTA/(Agx)6 and TNTA/(P/MQDs/P/Agx)6, which are used to evaluate the charge separation efficiency of the photoelectrodes. The PL peak originates from the recombination of photogenerated electron–hole pairs, and a lower peak intensity suggests higher efficiency of charge separation. As shown in Fig. 5d, TNTA/(P/MQDs/P/Agx)6 exhibits the lowest PL intensity, indicating that the cascade electron transport channel endowed by the synergy of PDDA and MQDs effectively promotes charge separation and migration over Agx@GSH NCs. As shown in Fig. 5e, TRPL results unveil that TNTA/(P/MQDs/P/Agx)6 shows the longest average electron lifetime (5.08 ns), which is higher than that of TNTA (2.14 ns) and TNTA/(Agx)6 (3.35 ns). This further validates the key role of the PDDA and MQDs in optimizing the electron migration pathway and suppressing carrier recombination.
According to the above analysis, the PEC water oxidation mechanism of the TNTA/(PDDA/MQDs/PDDA/Agy)n [Agy NCs = Agx@GSH, Ag9(GSH)6, Ag16(GSH)9] multilayer heterostructure photoelectrodes is proposed (Fig. 5f). Based on the DRS (Fig. S23a and b) and M–S results (Fig. S23c), conduction band (CB) and valence band (VB) potentials of TiO2 are determined to be −0.2 V and 2.87 V vs. NHE, respectively. Simultaneously, the highest occupied molecular orbital (HOMO) energy level of Agy NCs (y = x, 9, 16) was determined through CV measurement (Fig. S24–S26). Additionally, the lowest unoccupied molecular orbital (LUMO) energy levels of Agy NCs (Agx@GSH NCs: −1.03 V, Ag9(GSH)6 NCs: −1.85 V, Ag16(GSH)9 NCs: −2.01 V vs. NHE) were calculated based on their band gaps. In this way, energy band structures of Agy NCs and the TNTA substrate are determined, and simultaneously their energy level alignments are gained. Under visible light irradiation, the wide-bandgap TNTA substrate cannot be photoexcited, while the Agy NCs acting as the photosensitizer absorb photons and generate photogenerated electron–hole pairs. Notably, the LUMO levels of Agy NCs are more negative than the CB of TiO2 (−0.2 V vs. NHE), forming the energetically and kinetically favorable staggered energy level configuration that facilitates interfacial charge transfer. This enables the photogenerated electrons from Agy NCs to spontaneously migrate to the CB of TiO2. Furthermore, the synergistic electron-withdrawing effect triggered by the intermediate PDDA and MQD layers drives the high-efficiency and directional electron transfer from the Agy NCs to the TNTA substrate, ultimately enhancing the charge separation in Agy NCs. Then, electrons collecting in the CB of TiO2 transfer to the cathode via the external circuit to participate in the reduction reaction to generate hydrogen, while the holes in the HOMO level of Agy NCs are involved in the water oxidation reaction on the surface of the photoanode (Fig. S27). Overall, the synergy of PDDA and MQDs as concurrent electron-withdrawing mediators results in the formation of a cascade electron transport channel, which greatly promotes interfacial charge separation, enhancing the PEC water oxidation performances of TNTA/(PDDA/MQDs/PDDA/Agy)n multilayer heterostructures.
4 Conclusions
In summary, TNTA/(PDDA/MQDs/PDDA/Agy)n multilayer heterostructure photoanodes were exquisitely designed through a simple and green LbL assembly strategy under ambient conditions, by which tailor-made PDDA, MQDs and Agy NCs [Agx@GSH, Ag9(GSH)6, Ag16(GSH)9] with opposite charges were used as the building blocks and alternately assembled on the TNTA substrate. The unique spatially highly ordered interface configuration in the TNTA/(PDDA/MQDs/PDDA/Agy)n heterostructure photoanode as well as the synergistic electron-withdrawing capabilities of PDDA and MQDs afford a high-efficiency cascade charge transport channel and enable precise control over interfacial charge separation and migration over Agy NCs, considerably enhancing the PEC water oxidation performances. Our work provides conceptually inspiring ideas for the strategic modulation of charge transfer over metal NCs towards solar energy conversion.Author contributions
Peng Su and Jia-Liang Liu drafted the manuscript. Fang-Xing Xiao guided this work and corrected the manuscript. All the authors contributed to a critical discussion on the data and the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08806b.Acknowledgements
The support provided by the Program for Minjiang scholar professorship is gratefully acknowledged. This work was financially supported by the National Natural Science Foundation of China (No. 21703038 and 22072025) and Natural Science Foundation of Fujian province (2024J01263). Financial support from the State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, and Chinese Academy of Sciences is acknowledged (No. 20240018).References
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- M. A. Abbas, T. Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2015, 138, 390–401 CrossRef.
- Z. Q. Wei, S. Hou, X. Lin, S. Xu, X. C. Dai, Y. H. Li, J. Y. Li, F. X. Xiao and Y. J. Xu, J. Am. Chem. Soc., 2020, 142, 21899–21912 CrossRef CAS PubMed.
- Z. H. Zheng, F. X. Xiao and Y. J. Xu, Small, 2025, 21, 2505282 CrossRef CAS.
- L. C. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS.
- A. W. Cook and T. W. Hayton, Acc. Chem. Res., 2018, 51, 2456–2464 CrossRef CAS.
- P. Su, B. Tang and F. X. Xiao, Small, 2023, 20, 2307619 CrossRef PubMed.
- S. Hou, M. H. Huang and F. X. Xiao, J. Mater. Chem. A, 2022, 10, 7006–7012 RSC.
- Y. Zhang, C. L. Tan, Y. J. Xu and Z. R. Tang, Mol. Catal., 2026, 588, 115545 CAS.
- C. L. Tan, M. Y. Qi, Z. R. Tang and Y. J. Xu, Artif. Photosynth., 2025, 1, 144–155 CrossRef CAS.
- Z. P. Zeng, F. X. Xiao, X. C. Gui, R. Wang, B. Liu and Y. T. T. Thatt, J. Mater. Chem. A, 2016, 4, 16383–16393 Search PubMed.
- P. Su, S. Li and F. X. Xiao, Small, 2024, 20, 2400958 CrossRef CAS PubMed.
- L. F. Cai, Q. Chen, R. J. Xu, T. Li, J. Liang and F. X. Xiao, Inorg. Chem., 2024, 63, 24382–24391 Search PubMed.
- B. J. Liu, H. Liang, Q. L. Mo, S. Li, B. Tang, S. C. Zhu and F. X. Xiao, Chem. Sci., 2022, 13, 497–509 Search PubMed.
- S. Li, Q. L. Mo, S. C. Zhu, Z. Q. Wei, B. Tang, B. J. Liu, H. Liang, Y. Xiao, G. Wu, X. Z. Ge and F. X. Xiao, Adv. Funct. Mater., 2022, 32, 2110848 Search PubMed.
- H. Alijani, A. R. Rezk, M. M. Khosravi Farsani, H. Ahmed, J. Halim, P. Reineck, B. J. Murdoch, A. El-Ghazaly, J. Rosen and L. Y. Yeo, ACS Nano, 2021, 15, 12099–12108 Search PubMed.
- Q. Li, D. Shen, Z. Xiao, X. Liu, X. Xu, M. Wu, W. Wang, L. Liu, Q. Li and X. Li, Small, 2025, 21, 2411665 CrossRef CAS.
- Q. Xue, H. J. Zhang, M. S. Zhu, Z. X. Pei, H. F. Li, Z. F. Wang, Y. Huang, Y. Huang, Q. H. Deng, J. Zhou, S. Y. Du, Q. Huang and C. Y. Zhi, Adv. Mater., 2017, 29, 1604847 Search PubMed.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- R. Tang, S. J. Zhou, C. X. Li, R. Chen, L. Y. Zhang, Z. W. Zhang and L. W. Yin, Adv. Funct. Mater., 2020, 30, 2000637 Search PubMed.
- H. M. Wang, R. Zhao, H. X. Hu, X. W. Fan, D. J. Zhang and D. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 40176–40185 Search PubMed.
- Y. J. Li, L. Ding, Y. C. Guo, Z. Q. Liang, H. Z. Cui and J. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 41440–41447 Search PubMed.
- H. Liang, B. J. Liu, B. Tang, S. C. Zhu, S. Li, X. Z. Ge, J. L. Li, J. R. Zhu and F. X. Xiao, ACS Catal., 2022, 12, 4216–4226 Search PubMed.
- Y. S. Cai, J. Q. Chen, P. Su, X. Yan, Q. Chen, Y. Wu and F. X. Xiao, Chem. Sci., 2024, 15, 13495–13505 Search PubMed.
- Z. Y. Li, Y. H. Chen, J. R. Zhu, Q. Chen, S. J. Lu and F. X. Xiao, Inorg. Chem., 2023, 62, 16965–16973 Search PubMed.
- Y. Xiao, Q. L. Mo, G. Wu, K. Wang, X. Z. Ge, S. R. Xu, J. L. Li, Y. Wu and F. X. Xiao, J. Mater. Chem. A, 2023, 11, 2402–2411 Search PubMed.
- Q. L. Mo, J. L. Li, S. R. Xu, K. Wang, X. Z. Ge, Y. Xiao, G. Wu and F. X. Xiao, Adv. Funct. Mater., 2022, 33, 2210332 CrossRef.
- S. H. Xu, T. Gao, X. Y. Feng, Y. N. Mao, P. P. Liu, X. J. Yu and X. L. Luo, J. Mater. Chem. B, 2016, 4, 1270–1275 Search PubMed.
- Z. P. Zeng, Y. B. Yan, J. Chen, P. Zan, Q. H. Tian and P. Chen, Adv. Funct. Mater., 2018, 29, 1806500 CrossRef.
- F. X. Xiao and B. Liu, Nanoscale, 2017, 9, 17118–17132 Search PubMed.
- Z. B. Li, H. D. Bian, X. F. Xiao, J. D. Shen, C. H. Zhao, J. Lu and Y. Y. Li, ACS Appl. Nano Mater., 2019, 2, 7372–7378 CrossRef CAS.
- Z. Q. Wei and F. X. Xiao, Inorg. Chem., 2023, 62, 6138–6146 CrossRef CAS.
- S. Y. Wang, D. S. Yu and L. M. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS.
- S. Y. Yu and W. E. O’Grady, J. Electrochem. Soc., 2000, 147, 2952–2958 Search PubMed.
- V. Natu, M. Benchakar, C. Canaff, A. Habrioux, S. Célérier and M. W. Barsoum, Matter, 2021, 4, 1224–1251 Search PubMed.
- S. Kment, F. Riboni, S. Pausova, L. Wang, L. Wang, H. Han, Z. Hubicka, J. Krysa, P. Schmuki and R. Zboril, Chem. Soc. Rev., 2017, 46, 3716–3769 Search PubMed.
- P. Su, X. Yan and F. X. Xiao, Chem. Sci., 2024, 15, 14778–14790 RSC.
- Q. Q. Song, J. Q. Li, L. Wang, Y. Qin, L. Y. Pang and H. Liu, J. Catal., 2019, 370, 176–185 CrossRef CAS.
- Y. K. Quan, J. N. Li, X. Z. Li, R. X. Chen, Y. Z. Zhang, J. Y. Huang, J. Hu and Y. K. Lai, Appl. Catal. B Environ. Energy, 2025, 362, 124711 CrossRef CAS.
- Z. P. Zeng, Y. B. Li, S. F. Chen, P. Chen and F. X. Xiao, J. Mater. Chem. A, 2018, 6, 11154–11162 RSC.
- Z. Y. Li, M. Yuan and F. X. Xiao, Small, 2025, 21, 2409513 CrossRef CAS.
- S. Hou, Q. L. Mo, S. C. Zhu, S. Li, G. C. Xiao and F. X. Xiao, Inorg. Chem., 2021, 61, 1188–1194 CrossRef PubMed.
- X. Yan, M. Yuan, Y. L. Yuan, P. Su, Q. Chen and F. X. Xiao, Chem. Sci., 2024, 15, 10625–10637 RSC.
| This journal is © The Royal Society of Chemistry 2026 |