
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of solvent-free microwave nano-architectonics for the preparation of strongly coupled sub-nanometer interface materials: mechanisms, structures, and electrocatalytic applications
Yujia
Gao
a,
Wenxia
Xu
ab,
Hongdong
Li
a,
Jingqi
Chi
 a,
Jianping
Lai
*a and
Lei
Wang
*ab
a,
Jianping
Lai
*a and
Lei
Wang
*ab
aState Key Laboratory Base of Eco-Chemical Engineering, Ministry of Education, International Science and Technology Cooperation Base of Eco-chemical Engineering and Green Manufacturing, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: jplai@qust.edu.cn; inorchemwl@126.com
bShandong Engineering Research Center for Marine Environment Corrosion and Safety Protection, College of Environment and Safety Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
First published on 22nd December 2025
Abstract
Subnanometer materials, as an ideal platform for constructing strongly coupled interfaces at the atomic/cluster scale, are at the core of advancing efficient energy conversion technologies due to their ultimate atomic utilization efficiency, quantum confinement effects, and high specific surface area. The strongly coupled sub-nano interfacial structures developed based on such materials can simultaneously enhance the activity, selectivity, and durability of catalysts, holding the potential to overcome the limitations of traditional nanocatalysts in terms of atomic utilization and catalytic performance. However, the controllable synthesis of these materials remains challenging, constrained by the heavy reliance of traditional solvothermal methods on solvents, their high environmental footprint, and slow thermodynamic processes, making it difficult to capture metastable subnanometer structures. To address the controllable preparation of these materials, solvent-free microwave synthesis has emerged. This technique leverages the selective absorption of microwave energy by polar functional groups to generate transient high temperatures and ultrafast kinetics under solvent-free conditions, bypassing traditional thermodynamic limitations and providing a novel pathway for the precise synthesis of metastable subnanometer structures. This article systematically elaborates on the synthesis principles, research progress, and application prospects of such materials, aiming to provide theoretical support for the further development and industrialization of high-performance subnano catalytic materials.
1 Introduction
Precise structural regulation of catalytic materials is central to achieving efficient energy conversion and green chemical synthesis. Although traditional strongly coupled supported catalysts6–11 can enhance the stability and dispersion of active sites through strong coupling interactions, their active centers typically remain at the nanoscale, posing challenges for further improving atomic utilization efficiency. Sub-nanometer materials (SNMs),12–18 as a cutting-edge research direction, feature characteristic dimensions falling below the 10 nanometer range. This scale leads to a dramatic increase in the proportion of surface atoms, resulting in high atomic utilization, significant confinement effects, and a high specific surface area. These characteristics create an ideal platform for establishing robust metal–support electronic interactions at the atomic level. The strongly coupled sub-nano interfacial materials developed based on this premise, by constructing robust chemical bonds at the atomic/cluster scale, can not only significantly increase the density and stability of active sites but also optimize the adsorption energy barriers of reaction intermediates by precisely tailoring the interfacial electronic structure. This fine manipulation at the electronic scale holds the potential to simultaneously and markedly enhance the intrinsic activity, selectivity, and durability of catalysts, demonstrating significant potential in electrocatalysis23–29 and beyond that surpasses traditional nanomaterials (Fig. 1). | ||
| Fig. 1 Definition and advantages of strongly coupled sub-nanometer interface materials. | ||
However, the practical preparation of such materials still faces constraints, including insufficient precision in controlling subnanometer structures, heavy reliance on solvents, and high environmental impacts associated with conventional synthesis methods.31–33 To achieve controllable synthesis, novel techniques such as solvent-free microwave synthesis have emerged. This technique leverages the selective absorption of microwave energy by polar functional groups to achieve transient high temperatures and ultrafast kinetic processes under solvent-free conditions, effectively overcoming traditional thermodynamic limitations and providing a feasible pathway for the precise construction of metastable subnanometer structures. Based on this, strongly coupled sub-nano interfacial materials supported on various substrates, as well as metastable materials such as high-entropy alloys, defect-rich catalysts, unconventional phases, and immiscible systems, have demonstrated excellent performance advantages in electrocatalysis and other applications, laying a solid foundation for the development of next-generation high-performance catalytic materials.36–39
Our research group has achieved a series of advances in the design and preparation of novel catalytic materials in recent years, with a focus on developing various strongly coupled sub-nano interfacial materials possessing unique interfacial structures and electronic properties. In 2021, we successfully constructed a Ru–Mo2C@CNT strongly coupled sub-nano interfacial material.4 Through interfacial electronic restructuring, this material demonstrated excellent alkaline hydrogen evolution performance, achieving an overpotential as low as 15 mV at 10 mA cm−2 and operational stability exceeding 1000 h. In the same year, we developed a series of Ru–RE immiscible alloy materials (RE = Gd, Er, Yb, La), which exhibited outstanding catalytic performance due to their metastable structures formed via strong intermetallic electron coupling.19 In 2022, we overcame the technical bottleneck of noble metal cluster loading, developing highly loaded Pt/Pd/Ru subnanocluster functionalized carbon-based materials.3 The Ru/P–MoB catalyst developed in 2023, characterized by strong coupling interactions and an electron-enriched interface, achieved efficient and stable hydrogen evolution comparable to Pt/C across the entire pH range and even in seawater electrolysis.34 In 2025, we further constructed an Os–OsP2 heterointerfacial catalyst.49 By synergistically optimizing the water dissociation and hydrogen desorption processes, this catalyst significantly enhanced the hydrogen evolution performance and industrial application potential under wide-pH and seawater electrolysis conditions. The development of these material systems provides new insights for the design of high-efficiency energy conversion materials (Fig. 2).
 | ||
| Fig. 2 Historical development of solvent-free microwaves. Reproduced with permission.4 Copyright 2021, Nature Portfolio. Reproduced with permission.19 Copyright 2022, Elsevier. Reproduced with permission.34 Copyright 2023, Wiley. Reproduced with permission.41 Copyright 2023, Wiley. Reproduced with permission.43 Copyright 2024, Wiley. | ||
This article provides a systematic review of strongly coupled sub-nano interfacial materials prepared via solvent-free microwave synthesis technology, focusing on their unique advantages in atomic-level interface regulation and the construction of strong coupling interactions. Leveraging localized hot spot effects and ultrafast heating/cooling kinetics induced by selective microwave energy transfer enables the precise construction of non-equilibrium metastable materials, including strongly coupled sub-nano interfacial materials, high-entropy alloys, and defect-rich catalysts, among other metastable structures. By examining representative case studies, the structure–performance relationships between the interfacial structure and catalytic performance of these materials are thoroughly analyzed. Furthermore, the application potential and future development directions of such materials in the field of electrocatalysis are prospected (Scheme 1).
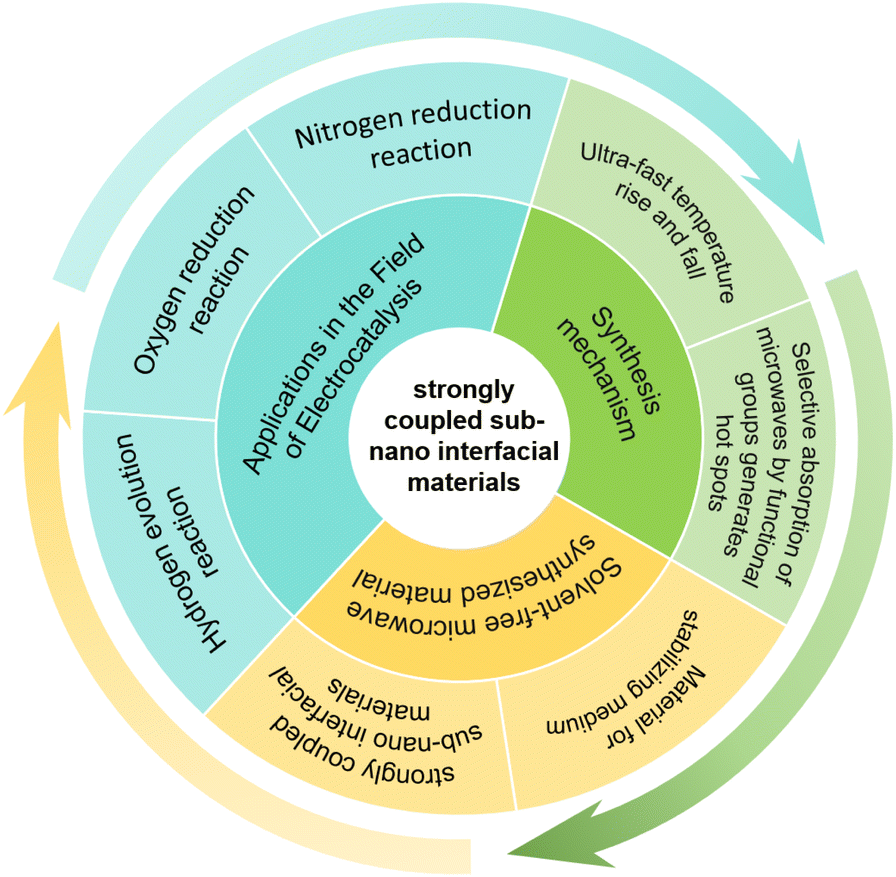 | ||
| Scheme 1 Mechanisms, synthetic materials and applications of solvent-free microwave synthesis technology. | ||
2 The core mechanism of solvent-free microwave synthesis
2.1 Functional group selective absorption of microwaves generates hot spots (which can form strongly coupled sub-nano interfacial materials)
The core mechanism of solvent-free microwave synthesis originates from the interaction between the microwave field and the dielectric properties of materials. Significant differences exist in the dielectric loss capabilities of various chemical functional groups towards microwave energy, and this selective absorption characteristic provides the physical basis for atomic-level interface regulation. As shown in Fig. 3, functional groups with strong polarity or conjugated structures (such as carbonyl and carboxyl groups) exhibit an “antenna effect” in the microwave field, preferentially absorbing microwave energy and forming transient high-energy “hot spots” at the molecular scale. Their thermodynamic behavior is significantly distinct from traditional uniform heating modes. | ||
| Fig. 3 Schematic mechanism of solvent-free microwave-induced functional group-selective hot-spot-driven strongly coupled catalyst synthesis. | ||
Taking the Ru–Mo2C@CNT heterojunction catalyst developed by our research group as an example,4 the polar functional groups on the surface of carboxylated multi-walled carbon nanotubes (CNTs) selectively absorb microwave energy, forming local high-temperature hot spots. These hot spots trigger the decomposition of metal carbonyl precursors (such as Ru3(CO)12 and Mo(CO)6) and drive the carbothermal reduction reaction using the CNT carbon source, thereby converting metal ions in situ into Ru0 and Mo2C. The microwave thermal effect simultaneously constructs an ultra-small Ru–Mo2C heterojunction with strong coupling interactions. Through this strong coupling and interfacial electron transfer, the subnanometer particles are firmly anchored, effectively addressing issues common in traditional methods, such as particle agglomeration, solvent pollution, and high energy consumption.
2.2 Ultra-fast heating and cooling (can form metastable materials)
The core mechanism by which solvent-free microwave synthesis enables the preparation of metastable materials lies in its utilization of instantaneous bulk energy input provided by microwave dielectric heating, combined with the inherent low heat capacity and low mass transfer resistance of the solvent-free system, achieving heating and cooling rates far exceeding those of traditional methods. This extremely non-equilibrium thermal environment breaks through the limitations of traditional thermodynamic equilibrium, allowing the entire process from precursor decomposition and rapid nucleation to structural freezing to be completed within a second-level time window. Atoms cannot fully diffuse and rearrange into the energy-minimized stable crystal structure within such a brief period, thus becoming “frozen” in a metastable state, forming unique microstructures such as high-density defects/vacancies, sub-nanometer scale grains, or amorphous–crystalline phase composites (Fig. 4). | ||
| Fig. 4 Solvent-free microwave ultrafast ramp-up/down synthesis of mesostable materials. | ||
Taking the WO3−x SNCs studied by our research group as an example,51 precisely controlled one-minute microwave irradiation triggers a sharp temperature increase in the system within a short duration. This rapid thermal increase prompts the rapid cleavage of the ammonium paratungstate (APT) precursor into W–O units, which uniformly disperse on the surface of the carbon carrier. Upon immediate termination of heating after precisely one minute of directional energy input, the system undergoes ultrafast cooling. This prevents the high-surface-energy sub-nanometer clusters (0.7 nm) from forming a stable crystalline phase through Ostwald ripening. Consequently, the non-stoichiometric WO3−x phase (W![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O = 1:2.71), rich in oxygen vacancies (Ov), is “frozen” in a metastable state. The simultaneously achieved ultrahigh loading of 49.29 wt% corroborates the efficient capture capability of ultrafast heating/cooling for thermodynamically unstable states, successfully balancing the conflict between the high density of active sites and structural stability inherent in metastable materials.
:O = 1:2.71), rich in oxygen vacancies (Ov), is “frozen” in a metastable state. The simultaneously achieved ultrahigh loading of 49.29 wt% corroborates the efficient capture capability of ultrafast heating/cooling for thermodynamically unstable states, successfully balancing the conflict between the high density of active sites and structural stability inherent in metastable materials.
3 Classification of materials synthesized by solvent-free microwave synthesis
3.1 Strongly coupled sub-nano interfacial materials
Strongly coupled sub-nanometer interface materials are centered on active units at the sub-nanometer scale (less than 10 nanometers), utilizing solvent-free microwave synthesis technology to establish robust chemical bonding or strong electron transfer interactions with the support at the atomic or cluster scale. This class of materials overcomes the limitations of conventional nanomaterials regarding the agglomeration of active components and electron transport efficiency, achieving synergistic optimization of active site density, electronic structure, and stability, and demonstrating outstanding performance in electrocatalysis and energy storage. The synthesis of these materials is centered on the local hotspot effects and ultrafast kinetics provided by the solvent-free microwave technique, offering an ideal pathway for atomic-level interface engineering.Subsequently, the high temperature in the hotspot region drives the rapid decomposition of metal precursors, generating ultrasmall metal/metal compound sub-nanometer active units with controllable sizes. Simultaneously, the polar functional groups on the carrier surface are activated at high temperature, reserving sufficient reactive sites for interface bonding. Under the synergistic effect of microwave thermal energy and the localized electric field, the sub-nanometer active units and the activated sites on the carrier further form chemical bonds such as M–O–C, M–P, and M–Ov–Ti (where Ov represents an oxygen vacancy), or establish strong electron transfer interactions through significant charge redistribution, ultimately constructing an atomically conjunct, strongly coupled interface. The entire process can be completed synchronously within 30–120 s, effectively circumventing the issue of weak interface coupling inherent in traditional “nucleation-first, loading-later” methods. This ensures that the material possesses high dispersion of active components, robust interfacial interactions, and efficient electron transport.
3.1.2.1 Carbon nanotubes. In the carrier system for strongly coupled sub-nanometer interface materials,1,52,53 carbon-based materials, with their one-dimensional, two-dimensional, and three-dimensional diversified microstructures, excellent microwave response characteristics from selective microwave absorption by surface polar functional groups, and high conductivity, have become the core carrier for solvent-free microwave synthesis. Their core advantages lie in: surface defect sites and oxygen-containing functional groups such as carboxyl and hydroxyl groups can act as microwave energy capture centers, forming local high-temperature hotspots within the microwave field. This simultaneously drives metal precursor reactions and the construction of strong interfacial coupling. Furthermore, carbon structures of different dimensions can adapt to various application scenarios such as high current density and flexible devices, providing flexible space for material function customization. The following section combines typical carbon-based carriers such as carbon nanotubes,54–57 graphene,58–61 carbon cloth,62–66 and Ketjen black to explain the formation mechanism of metastable phase structures when constructing strongly coupled sub-nanometer interface materials.
In specific material systems, carbon nanotubes (CNTs) serve as representative one-dimensional carriers. Their hollow tubular structure combines high conductivity and mechanical strength, while surface defects and carboxylation modifications further enhance their microwave absorption capability. For example, in the synthesis of Ru–Mo2C@CNT,4 the –COOH functional groups on the surface of carboxylated multi-walled carbon nanotubes selectively absorb microwave energy, forming localized ultra-high-temperature hotspots. This promotes the rapid decomposition of the Ru3(CO)12 and Mo(CO)6 precursors, generating ultrasmall Ru–Mo2C heterojunctions with an average size of 3.5 nm. The ultrafast heating kinetics of microwaves limit sufficient atomic diffusion, confining the heterojunction to the sub-nanometer scale. Simultaneously, the CNT surface firmly anchors it through covalent bonding and electron transfer interactions, forming a dense carbon layer coating structure. This successfully “freezes” the metastable heterojunction (Fig. 5a).22,67,68
 | ||
| Fig. 5 (a) High-resolution XPS spectrum of C 1s of CNTs and Ru–Mo2C@CNT. Reproduced with permission.4 Copyright 2021, Nature Portfolio. (b) RGO film with numerous functional groups before microwave irradiation. A magnified view depicts the rotating dipole of the RGO functional groups during microwave irradiation. Reproduced with permission.21 Copyright 2019, Cell Press. (c) Calculated OH-binding energies (ΔEOH) and H binding energies (ΔEH) for the Pt site, Nb site in PtNb, and Nb site in Nb2O5 (inset shows the model of PtNb–Nb2O5). Reproduced with permission.40 Copyright 2022, Elsevier. (d) Free-energy barriers of different catalysts, and the inset shows the crystal structures. Reproduced with permission.42 Copyright 2021, Wiley. (e) H adsorption energy of TiO2, Pt/TiO2, Pt/TiO2–OV and Pt/graphene. Reproduced with permission.41 Copyright 2023, Wiley. (f) Work functions of MoB and P–MoB. Reproduced with permission.34 Copyright 2024, Wiley. (g) Projected density of states of the Ru atom in RuP2 for halogen element (X = Cl, Br, and I)-modified Ru-based nanosheets. Reproduced with permission.46 Copyright 2024, Wiley. (h) Illustration of the optimization model of Ru@p-Co3HHTP2. Reproduced with permission.48 Copyright 2022, Elsevier. | ||
Two-dimensional graphene carriers are known for their atomic-level flatness derived from the sp2-hybridized carbon network. The selective absorption of microwave energy by edge defects and oxygen-containing functional groups (such as hydroxyl and epoxy groups) is even stronger, facilitating the construction of a more uniform strongly coupled interface. The team of Liangbing Hu21 utilized 300 °C-pretreated reduced graphene oxide (RGO) as a carrier. The enriched oxygen-containing functional groups on its surface selectively absorb energy via dipole rotation effects in a 2.45 GHz microwave field, forming a localized transient high-temperature field reaching up to 1600 K within 100 ms (Fig. 5b). This thermal field promotes the rapid nucleation of CoS. However, due to the extremely fast heating rate and ultrashort reaction time, the CoS atoms cannot fully agglomerate to form a stable crystalline phase. Consequently, they are uniformly distributed on the RGO surface in the form of sub-nanometer particles, forming a strongly coupled interface with lattice coherence with RGO. After the microwave stops, the system cools rapidly, “freezing” this metastable structure. The electronic reconstruction at the interface induces a strong coupling effect, significantly optimizing charge transport and catalytic stability. This mechanism can also be extended to various metal systems such as Ru, Pd, and Ir.69
Three-dimensional flexible carrier carbon cloth (CC) possesses a three-dimensional interconnected conductive network and a hierarchical pore structure, demonstrating significant advantages in high-current-density electrolysis and flexible device applications. The oxygen-containing functional groups on its fiber surface can enhance the metal–support interfacial coupling, while the three-dimensional pores provide physical confinement space for the active components. Taking the PtNb–Nb2O5/CC material synthesized by our group40 as an example, acid pretreatment increases the density of oxygen-containing functional groups on the carbon cloth surface. In the microwave field, these functional groups selectively absorb microwaves, generating transient hot spots that drive the rapid decomposition of the precursors. The kinetic limitations of microwave heating prevent Pt and Nb atoms from fully diffusing to form a stable crystalline phase, ultimately resulting in the formation of Pt3Nb–Nb2O5 sub-nano heterojunctions with an average particle size of 2.5 nm. The electron transfer effect mediated by the functional groups on the carbon cloth fiber surface anchors these heterojunctions. After the microwave stops, the system cools rapidly, “freezing” the strong coupling between the heterojunctions and the support, thereby locking the metastable structure (Fig. 5c). Additionally, the three-dimensional flexible structure of the carbon cloth enhances the local electric field through fiber microcavity resonance and dynamically regulates the interfacial stress via its thermal expansion characteristics, further maintaining the stability of the metastable structure. This enables the material to operate stably at high current densities of 500 mA cm−2 for extended periods.
Ketjen black (KB) is distinguished by its ultra-high specific surface area and extremely abundant surface oxygen-containing functional groups, which serve as its primary advantages. Its surface functional groups act as high dielectric loss units, capable of rapidly inducing the formation of localized hotspots in solvent-free systems. In the preparation of the Pt61La39@KB material by our research group,42 the oxygen-containing functional groups on the KB surface absorb microwave energy to form hotspots, driving the rapid reaction of Pt(acac)2 and La(acac)3 precursors. Due to the ultrafast heating of the hotspots, Pt and La atoms cannot diffuse sufficiently to form a stable crystalline phase, resulting in the generation of Pt–La alloy sub-nanometer particles with an average size of 3 nm. After the microwave stops, the system cools rapidly, and the particles are anchored onto the KB surface via a Pt–O–C covalent bond network, “freezing” into a metastable structure. The three-dimensional mesoporous structure of KB enhances the localization of microwave energy through multiple scattering effects, further suppressing atomic diffusion. Meanwhile, the injection of 4f electrons from lanthanum causes a downshift in the d-band center of platinum, synergistically optimizing the electronic structure and maintaining the stability of the metastable structure (Fig. 5d).
The common mechanism for the formation of metastable phases in carbon-based carriers when constructing strongly coupled sub-nanometer interface materials lies in: relying on the microwave selective absorption characteristics of polar functional groups such as carboxyl and hydroxyl to generate local high-temperature hotspots, rapidly driving the reaction of metal precursors; the ultrafast heating of microwaves and short reaction time strictly limit the long-range diffusion of atoms, forcing the metal components to exist at the sub-nanometer scale; the carriers tightly anchor the sub-nanometer particles through covalent bonding, electron transfer, and other interactions; after the microwave stops, the system rapidly dissipates heat, “freezing” the strong coupling interaction between the sub-nanometer particles and the carriers, thereby successfully locking the metastable structure. Carbon structures of different dimensions, by adapting to specific application scenarios, further ensure the stability of the metastable structure under real working conditions, effectively solving problems such as particle agglomeration and solvent dependence in traditional synthesis methods, laying a solid foundation for the industrial development of such high-performance materials.
3.1.2.2 Metal oxide supports. Metal oxide carriers exhibit unique advantages in constructing strongly coupled sub-nanometer interface materials. Their tunable surface oxygen vacancies (Ov), abundant coordination sites, and potential strong electronic interactions with active components provide an ideal platform for atomic-level precise interface construction. Compared to carbon-based carriers, the oxygen vacancies and polar functional groups like hydroxyls on the surface of metal oxides possess a stronger selective absorption capability for microwave energy, enabling more efficient formation of localized high-temperature hotspots. This facilitates the highly dispersed anchoring of active sites and the regulation of the interface electronic structure under solvent-free conditions.
The Pt–Ov–TiO2 strongly coupled sub-nanometer interface material constructed by our research group is a typical representative of this class of materials.41 The formation of its metastable structure critically relies on the kinetic control mechanism provided by the solvent-free microwave synthesis technique. Based on dry mechanically mixed precursors, the microwave field preferentially acts on the polar functional groups on the TiO2 surface, generating instantaneous high-temperature microzones. Within this non-equilibrium thermal environment, the reductive cleavage of Pt–Cl bonds and the in situ generation of Ov in the TiO2 lattice proceed simultaneously. Reduced Pt atoms, constrained by limited diffusion in the high-temperature environment, cannot migrate sufficiently to form a thermodynamically stable crystalline phase. Consequently, they are locked at a sub-nanometer scale (less than 1 nanometer) and firmly anchored to the carrier surface via a strongly coupled Pt–Ov–Ti interface. XPS analysis confirms significant electron transfer at the interface, indicated by a negative shift of 0.3 eV in the Pt 4f binding energy. DFT calculations further reveal a 63% increase in the adsorption energy of Pt. These optimizations in the electronic structure effectively suppress the migration and agglomeration of Pt atoms (Fig. 5e). The entire synthesis process requires only 1 minute of microwave irradiation and is completed under mild reducing conditions at 80 °C. These gentle reaction conditions effectively avoid the crystal phase relaxation and particle growth issues associated with conventional high-temperature processes, thereby successfully “freezing” the metastable structure.
The core mechanism of this synthesis strategy exhibits excellent universality and can be further extended to other metal oxide carrier systems such as CeO2 and ZrO2. By regulating the surface hydroxyl density and lattice oxygen mobility of the carriers, active interfaces with specific electronic structures can be directionally designed, providing a feasible technical pathway for the development of diversified high-performance catalytic materials.
3.1.2.3 Metal phosphide supports. Metal phosphide carriers, with their unique electronic structure, high conductivity, and strong electronic coupling effects with active components, have become an ideal platform for constructing high-efficiency supported catalysts. Compared to traditional carriers, functional groups such as P–H and P
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O on the surface of metal phosphides exhibit excellent selective absorption capability for microwave energy. In solvent-free systems, these groups can form transient high-temperature hotspots through dielectric loss. This characteristic enables the simultaneous in situ anchoring of metal sub-nanometer particles and interface reconstruction, achieving strong coupling regulation at the atomic scale. It also optimizes water dissociation kinetics and the adsorption behavior of reaction intermediates, providing highly efficient and stable catalytic interfaces for energy conversion reactions such as the hydrogen evolution reactions (HER) and oxygen evolution reactions (OER).
O on the surface of metal phosphides exhibit excellent selective absorption capability for microwave energy. In solvent-free systems, these groups can form transient high-temperature hotspots through dielectric loss. This characteristic enables the simultaneous in situ anchoring of metal sub-nanometer particles and interface reconstruction, achieving strong coupling regulation at the atomic scale. It also optimizes water dissociation kinetics and the adsorption behavior of reaction intermediates, providing highly efficient and stable catalytic interfaces for energy conversion reactions such as the hydrogen evolution reactions (HER) and oxygen evolution reactions (OER).
Leveraging the above advantages, our research group developed various types of strongly coupled sub-nanometer interface materials using solvent-free microwave synthesis technology and achieved precise construction of metastable structures through differentiated kinetic control strategies. In the Ru/P–MoB system,34 using MoB, RuCl3, and NaH2PO2·H2O as precursors, microwave-induced plasma hotspots created an instantaneous high-energy environment within 60 s, simultaneously achieving the in situ phosphidation of MoB and the reduction of Ru3+ to 2 nm ultrasmall clusters. Due to kinetic limitations, the Ru clusters cannot form a stable crystalline phase but are anchored onto the P–MoB surface via Ru–Mo and Ru–P bonds, forming a strongly coupled metastable structure. Interfacial electron redistribution significantly optimizes the carrier's work function (Fig. 5f), while the ultra-short reaction time effectively prevents crystal phase relaxation and simultaneously improves water molecule adsorption and water dissociation performance.
In the design of the halogen-doped Ru/RuP2 system,46 our group innovatively introduced halogen salts as regulatory units. Using RuCl3·xH2O, NaH2PO2·H2O, and halogen salts as precursors, microwave phosphide plasma simultaneously completed the reduction of Ru3+, the formation of the RuP2 carrier, and the in situ doping of halogen within 60 s. Ru sub-nanometer particles and RuP2 rapidly form a heterostructure, which is locked into a metastable state due to kinetic constraints. Among these, halogen precisely regulates the interface coupling strength through electron transfer effects. Particularly, the Br element, with its moderate electronegativity, optimizes the d-band center of Ru. Meanwhile, Ru–P and Ru–Ru bonds collectively lock the high-energy structure. The Br-induced electron redistribution maintains structural stability while significantly weakening the Ru–H binding energy (Fig. 5g).
For the Ru/WP material system,70 our research group (75) innovatively adopted a solvent-free microwave quasi-solid-state synthesis route. By constructing a quasi-solid-state system from Ru-based and WP precursors, and leveraging its low heat capacity and low mass transfer resistance, polar functional groups selectively absorb energy in the microwave field to form local hotspots, rapidly driving the reduction of the Ru precursor and the phosphidation process to form WP. The ultrafast heating characteristic of microwaves effectively limits atomic diffusion, maintaining the Ru particles at the sub-nanometer scale, while the WP carrier retains abundant metastable defects. The system's rapid cooling “freezes” the strong interaction between Ru and WP, forming a unique sponge-like porous metastable structure. This structure maintains stability through strong electronic coupling and spatial confinement effects. It not only possesses a high specific surface area of 14.84 m2 g−1 exposing more active sites but also significantly optimizes the catalytic reaction kinetics.
In summary, solvent-free microwave synthesis technology utilizes functional group-selective microwave absorption to form localized hotspots, combined with plasma-assisted or quasi-solid-state pathways. This approach solves the problems of high energy consumption, pollution, and particle agglomeration associated with traditional phosphidation processes, successfully constructing various strongly coupled sub-nanometer materials and optimizing catalytic performance. The tunable electronic structure and defect engineering of metal phosphide carriers also provide a sustainable solution for the low-cost, high-performance development of industrial-grade hydrogen evolution catalysts.
3.1.2.4 MOF supports. In the field of electrocatalytic hydrogen evolution, developing efficient and stable supported catalysts is a core challenge. Although traditional conductive metal–organic frameworks (MOFs) possess high conductivity and abundant active sites, their low catalytic activity and weak stability severely limit practical applications. To address this, the solvent-free microwave synthesis strategy has been developed. This strategy leverages the selective absorption of microwave energy by MOF functional groups to construct strong coupling between the carrier and metal sub-nanometer particles, providing an innovative direction for the design of high-performance catalysts.
The strongly coupled sub-nanometer interface material Ru@p-Co3HHTP2,48 constructed by our research group based on this strategy, has its formation mechanism for the metastable phase structure centered on the synergistic kinetic control between the MOF carrier and microwave energy: using conductive Co3HHTP2 MOF as the carrier and Ru3(CO)12 as the metal precursor, after solid-state grinding and mixing, the oxygen-containing functional groups (phenolic hydroxyl and carboxyl groups) and conjugated aromatic ring structures on the MOF surface, by virtue of their high dielectric loss properties, selectively absorb energy in the microwave field to form localized high-temperature hotspots. Within the ultrafast kinetic process under 60 seconds of microwave irradiation, the hotspots simultaneously trigger three key reactions: the rapid reduction of Ru3(CO)12 to approximately 5 nm Ru sub-nanometer particles (which cannot diffuse to form a stable crystalline phase due to the extremely short reaction time), the controllable partial carbonization of the Co3HHTP2 surface forming a metastable amorphous carbon layer, and the reconstruction of a strong coupling interaction between Ru and the MOF via Ru–O–Co bonds (Fig. 5h). The rapid cooling of the system after microwave cessation “freezes” these interactions, locking the sub-nanometer metastable structure. Meanwhile, strong interfacial electron transfer (evidenced by a negative shift in the Ru 3p binding energy) enhances the interaction between Ru and the carrier, while the spatial confinement effect of the amorphous carbon layer inhibits the growth of Ru particles; together, they maintain the stability of the metastable structure. Ultimately, this enables the material to exhibit excellent alkaline hydrogen evolution performance and provides a kinetic control paradigm for the preparation of MOF-based strongly coupled sub-nanometer materials.
This research not only confirms that the solvent-free microwave method can precisely regulate the synergistic effect between carrier carbonization and metal loading but also breaks through the bottlenecks in the stability and activity of MOF carriers through strong coupling effects, providing a universal paradigm for the green synthesis of strongly coupled catalytic systems. Future work could further explore the microwave response mechanisms of diverse MOF ligands to expand the application scenarios of such materials.
3.2 Metastable materials
Metastable materials refer to non-equilibrium material systems formed through the ultrafast heating/cooling kinetics of solvent-free microwave synthesis. These materials exist in a high-energy state thermodynamically; however, the kinetic barriers constructed by rapid cooling nearly halt atomic diffusion, thereby “freezing” the high-energy metastable structure and allowing it to remain stable over a long period at ambient temperature and pressure. Their core value lies in breaking the limitations of traditional thermodynamic synthesis. Leveraging the unique electronic configurations conferred by their non-equilibrium structures, they demonstrate performance advantages in fields such as electrocatalysis and energy storage that are unmatched by conventional stable-phase materials.After the reaction terminates, the low heat capacity characteristic of the solvent-free system causes the temperature to drop rapidly to room temperature, halting atomic movement almost instantaneously. This rapidly “freezes” the high-energy metastable nuclei and structures. This step is key to the formation of the metastable structure. For example, in immiscible Ru–RE alloys, rapid quenching can inhibit element segregation, locking in the atomically mixed structure; in WO3 rich in oxygen vacancies, rapid cooling prevents the filling of vacancies by oxygen atoms, maintaining the non-stoichiometric state. Concurrently, the localized electric field of the microwave can also assist in stabilizing the metastable phases. It can promote defect generation or induce the nucleation of unconventional crystal phases, and combined with mechanisms such as carrier confinement and elemental synergy, further enhances the kinetic stability of the structure.
3.2.2.1 Defect-rich catalysts. Defect-rich catalysts, as an important category in metastable material systems, demonstrate significant advantages in the field of catalysis due to their unique high-density and tunable defect structures. By precisely constructing various defect types such as lattice distortion, heterointerfaces, oxygen vacancies, and atomic vacancies, these materials can not only directly participate in catalytic reactions as active centers but also optimize the electronic structure of active sites and the adsorption behavior of reaction intermediates through electronic reconstruction effects, thereby breaking through the performance limitations of traditional materials in catalysis.
Solvent-free microwave synthesis technology is a core method for constructing such materials. Its common mechanism revolves around “instantaneous high temperature inducing defect formation – rapid cooling locking the metastable state” and is adaptable to diverse carrier systems. On carbon-based carriers,2 the instantaneous high temperature formed within 60 s of microwave irradiation drives the simultaneous phosphidation, reduction, and self-assembly of precursors, suppressing element volatilization and particle agglomeration. The subsequent ultrafast cooling “freezes” the heterointerface formed between Ru and Ru2P into a metastable state (Fig. 6a). Noble metal doping can further induce lattice distortion through electronic synergy, enhancing structural stability and catalytic activity. Extending to vacancy defect systems, microwave instantaneous high temperature promotes the formation of intermetallic compound nuclei from Pd and M (Pb, Sn, and In) precursors.5 By controlling the irradiation time, the vacancy concentration can be directionally regulated. The quenching effect then inhibits atomic rearrangement, stabilizing the thermodynamically unstable ordered vacancy structure within the lattice (Fig. 6b).
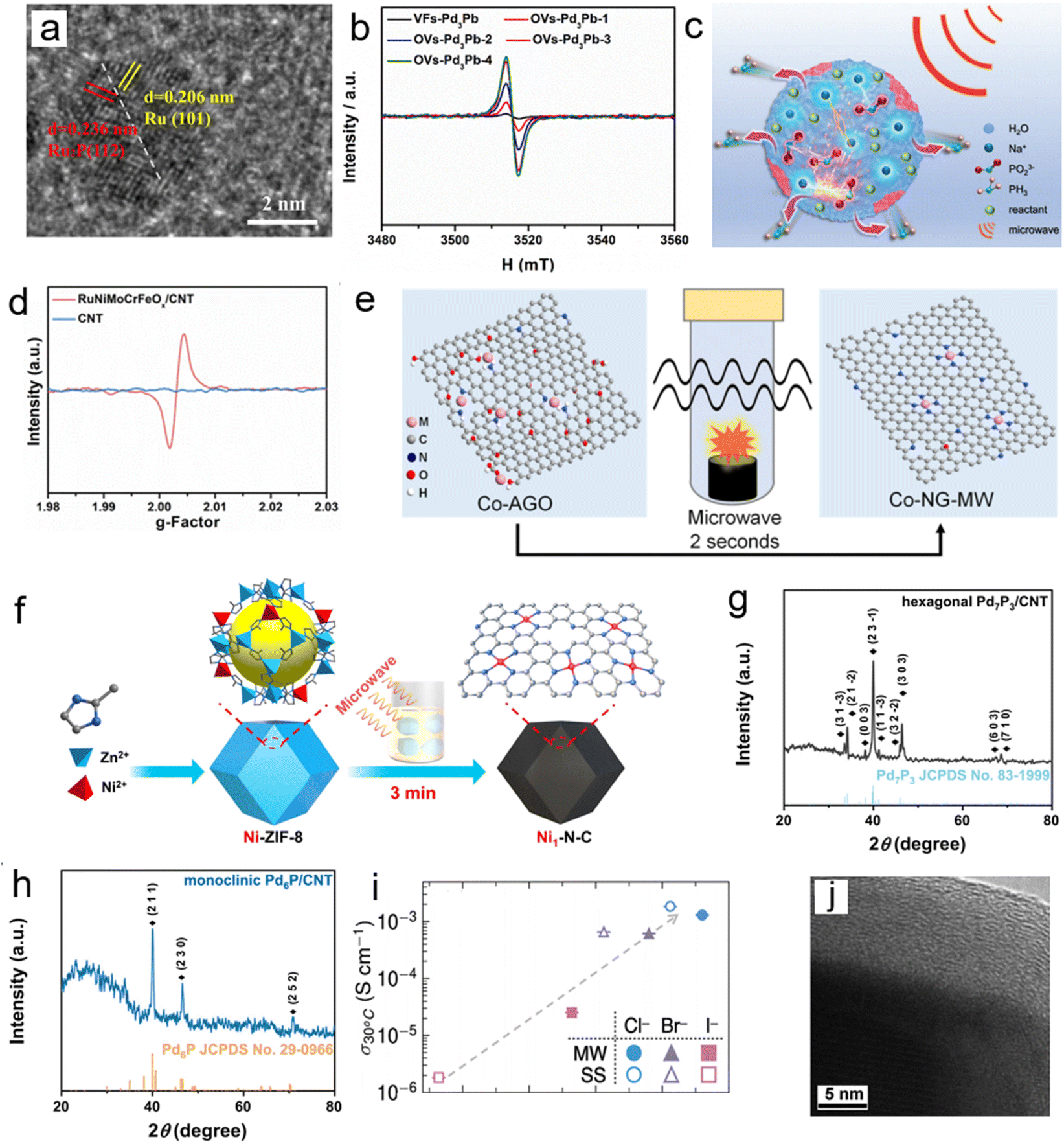 | ||
| Fig. 6 (a) HRTEM images of Pt0.095−Ru2P@Ru/CNT. Reproduced with permission.2 Copyright 2021, American Chemical Society (b) Electron spin resonance (ESR) spectroscopy of the intermetallic VFs-Pd3Pb and the intermetallic OVs-Pd3Pb-x (x = 1, 2, 3, 4). Reproduced with permission.5 Copyright 2022, Elsevier. (c) Mechanism of the microwave reaction. Reproduced with permission.35 Copyright 2024, Wiley. (d) The ESR spectra of RuNiMoCrFeOx/CNT and CNT catalysts before the OER. Reproduced with permission.20 Copyright 2022, RSC. (e) Schematic illustration of the preparation route to Co-NG-MW. Reproduced with permission.44 Copyright 2018, Wiley. (f) Illustration showing the fabrication of Ni1–N–C by microwave-assisted rapid pyrolysis. Reproduced with permission.43 Copyright 2024, Wiley. (g) XRD pattern of hexagonal Pd7P3/CNT. (h) XRD pattern of monoclinic Pd6P/CNT. Reproduced with permission.45 Copyright 2022, Elsevier. (i) Ionic conductivity at T = 30 °C plotted against median PS43− displacement. Reproduced with permission.47 Copyright 2025, Wiley. (j) HRTEM images of the edge of the CoMoO4 crystallite. Reproduced with permission.50 Copyright 2012, RSC. | ||
In the direction of oxide carriers, the formation of metastable PtRu alloy materials supported on WO3 relies on the transient severe oxidation of H2PO2− under the synergy of electric and microwave fields,35 creating a local high-temperature microzone that drives the formation of oxygen vacancies in WO3 and the alloying of PtRu (Fig. 6c). Subsequent rapid cooling prevents oxygen vacancy filling and alloy agglomeration, locking the high-energy metastable interface.
For low-loading Ru–Pt metastable materials on phosphorus-doped black titanium ore (P–TiO2−x) carriers,71 microwave instantaneous high energy simultaneously triggers the formation of oxygen vacancies in TiO2 and the alloying of Ru–Pt. The ultra-fast cooling effect inhibits atomic diffusion, freezing the mixed-phase structure and lattice-distorted heterojunction. Furthermore, phosphorus doping and oxygen vacancies synergistically induce electronic reconstruction, enhancing structural stability and optimizing interfacial electron transport kinetics.
In summary, solvent-free microwave synthesis technology, leveraging ultrafast heating/cooling kinetics and synergistic functional group energy absorption, can precisely regulate multiple types of defects and is adaptable to various carriers like carbon-based materials, oxides, and phosphides. It provides an efficient and green synthesis pathway for developing high-performance metastable catalytic materials.
3.2.2.2 High-entropy materials. Although high-entropy materials possess unique catalytic potential due to multi-element synergistic effects, their application is limited by element segregation and phase separation often caused by the high-temperature, prolonged processes of traditional synthesis. Solvent-free microwave synthesis technology, leveraging its ultrafast heating/cooling kinetic characteristics, provides an innovative pathway for the precise construction of carbon nanotube-supported high-entropy oxides. The rapid quenching effect freezes the non-equilibrium structure, achieving atomic-level mixing of multiple elements and synergistic control of defects, opening a new direction for the controllable preparation of high-entropy materials.
Based on this, the metastable RuNiMoCrFeOx high-entropy oxide material developed by our research group20 relies on a “ultrafast heating – rapid quenching and locking” kinetic mechanism for the formation of its metastable phase structure: using Ru, Ni, Mo, Cr, and Fe metal salts as precursors, which are solid-state ground with carbon nanotubes into a uniform mixture, 120 seconds of microwave irradiation constructs a transient high-temperature field, driving the synchronous reduction of multiple metal ions and atomic diffusion. The subsequent rapid quenching significantly reduces the atomic diffusion rate, suppressing element segregation and lattice relaxation, and kinetically locks the five elements into a single solid solution phase, forming uniformly sized (approximately 6 nm) sub-nanometer particles. Furthermore, the synergistic effect of transient high temperature and rapid quenching induces oxygen vacancy defects (Fig. 6d), enhancing the material's unique electronic properties and laying the foundation for its catalytic performance.
This mechanism breaks through the limitations of traditional thermodynamic equilibrium, simultaneously achieving stabilized high configurational entropy and the construction of oxygen vacancy defects. It also offers advantages such as being solvent-free, having rapid reaction times, and good scalability, providing a green and efficient solution for the design of carbon nanotube-supported high-entropy materials. In the future, this technology is expected to be extended to diverse carrier systems, providing technical support for the development of high-performance catalytic materials.
3.2.2.3 Single-atom catalysts. Single-atom catalysts (SACs) demonstrate significant potential in the field of catalysis due to their maximized atom utilization efficiency and unique electronic structural properties. However, traditional synthesis methods, such as solvothermal methods, often face challenges like migration and agglomeration of metal atoms and weak carrier interface coupling. Solvent-free microwave synthesis technology, leveraging its distinctive ultrafast heating/cooling kinetics and functional group-mediated localized hotspot effects, has successfully overcome these limitations, providing an innovative solution for the precise preparation of single-atom catalysts.
In the study of graphene-based single-atom catalysts, the M-NG-MW (M = Co/Ni/Cu) material developed by Imran Shakir's team exemplifies an advanced synthesis strategy.44 The team adopted a mechanism of “precursor anchoring – microwave ultrafast defect construction – single-atom locking” (Fig. 6e): first, atomic-level dispersed anchoring sites were established through the pre-coordination of metal ions with the amino groups of amine-functionalized graphene oxide (AGO). After freeze-drying, the solid precursor underwent only 5 seconds of microwave irradiation at 1000 W power. The instantaneous high temperature simultaneously completed the deoxygenation of graphene oxide, nitrogen doping, and the generation of carbon vacancy defects. The ultrafast heating and cooling rates effectively suppressed the thermal diffusion of metal atoms, promoting the formation of stable M–Nx–C active centers where metal species coordinate with pyridinic nitrogen.
The team of Hai-Long Jiang43 further expanded this technical pathway by innovatively developing a molten salt-mediated microwave synthesis strategy (Fig. 6f). By using a ZnCl2/KCl mixed molten salt in an air environment to form a liquid phase barrier, they achieved stable carbonization of Ni-ZIF-8 without the need for inert gas protection. The molten salt system significantly improved the conversion efficiency of microwave energy, and the ultra-high temperature field combined with extremely rapid heating/cooling kinetics effectively suppressed the agglomeration of nickel atoms. It is particularly noteworthy that the high-temperature environment simultaneously triggered the directional decomposition of the MOF framework, synchronously forming the nitrogen-doped carbon carrier, carbon vacancy defects, and Ni–N4 active centers. Furthermore, the activation of the carbon skeleton by K+ ions generated a mesoporous structure with a high specific surface area, significantly enhancing the mass transfer efficiency of the material.
These research findings fully demonstrate that solvent-free microwave synthesis technology, by precisely matching the microwave absorption characteristics of polar functional groups on the carrier surface with ultrafast kinetic processes, successfully achieves the localized reduction of metal atoms and their anchoring at defect sites. This technology not only addresses the stability challenges in the synthesis of single-atom catalysts but also opens new directions for the precise regulation of the electronic structure of catalysts.
3.2.2.4 Immiscible materials. Although immiscible material systems (such as rare earth-transition metal alloys) possess unique potential in the field of catalysis, their synthesis is limited by phase separation issues induced by the high oxygen affinity of the elements. Traditional methods (e.g., solvothermal methods) also struggle to overcome the reliance on reduction potential matching and the thermodynamic immiscibility constraint. Solvent-free microwave synthesis technology, leveraging its unique kinetic control mechanism, provides a breakthrough solution for the precise construction of such metastable materials.
The metastable Ru–RE/rGO (RE = Gd/Er/Yb/La) alloy material constructed by our research group19 has the formation of its metastable phase structure centered on a kinetic control mechanism of “selective localized ultra-high temperature – second-level rapid quenching and locking”: the microwave field selectively excites the oxygen-containing functional groups of the precursors, creating a transient localized ultra-high temperature field. This triggers the cleavage of the carbonyl ligands in Ru3(CO)12 and drives carbothermal reduction, achieving the simultaneous co-reduction of Ru3+ and RE3+. The second-level rapid quenching significantly reduces the atomic diffusion rate, strongly suppressing the thermodynamic segregation tendency of the two elements, thereby locking the inherently immiscible Ru and RE into an atomically uniform metastable alloy structure and avoiding their transformation into low-entropy stable phases. Concurrently, the strong electronic coupling (rGO → Ru electron transfer) between the rGO and the alloy further enhances the stability of the metastable state, solving the phase separation problem caused by the high oxygen affinity of the rare earth elements. This method enables gram-scale mass production (yield 82.1%) and, through the optimization of the interfacial electronic structure, endows the material with excellent alkaline hydrogen evolution performance, providing a new strategy for the preparation of immiscible metastable alloys and their application in energy-related catalysis.
3.2.2.5 Unconventional phases. Solvent-free microwave synthesis technology, leveraging the synergistic effects of ultrafast heating/cooling kinetics and transient high-temperature fields, breaks through the limitations of traditional thermodynamic equilibrium. Through the localized energy focusing characteristic of microwave dielectric heating, it suppresses the nucleation of stable phases within a time window of seconds, and then locks the high-energy metastable structures via a rapid quenching effect, providing an innovative method for the design of high-performance catalytic and energy materials.
In the phosphorus source regulation strategy developed by our research group,45 the precise synthesis of metastable metal phosphide materials was successfully achieved through an innovative mechanism of “phosphorus source directional regulation – microwave instantaneous alloying – kinetic phase locking”. After solid precursors were mixed by dry grinding, irradiation at 700 W for 75 s triggered energy focusing, generating millisecond-level ultrafast heating/cooling kinetics that drove the rapid alloying of metal and phosphorus elements. By precisely selecting sodium hypophosphite or sodium pyrophosphate as the phosphorus source, different crystalline phase structures such as hexagonal Pd7P or monoclinic Pd6P could be directionally prepared (Fig. 6g and h). The ultra-short reaction time effectively inhibited particle agglomeration, forming sub-nanometer particles with sizes of 4–5 nm. The electron transfer effect between phosphorus and metal elements further enhanced the stability of the metastable structure.
In the research on two-dimensional materials and composite systems, the team of Ilia Valov72 successfully constructed two types of metastable materials: noble metal phosphides (MPx/CNT, M = Pd, Pt, Ru) and molybdenum-based 2D materials through a synergistic strategy of “transient ultra-high temperature – ultrafast cooling”. In the synthesis of the MPx system, the localized ultra-high temperature field constructed by microwave irradiation, combined with differentiated regulation of the phosphorus source, directionally guided the crystal growth pathway; the ultrafast cooling process effectively inhibited element segregation, successfully locking the metastable crystalline phase. Particularly noteworthy is the synthesis of the molybdenum-based 2D materials: graphene-mediated localized microwave absorption created a unique thermal gradient and strong vertical electric field, which triggered the breaking of Mo–S bonds in MoS2 at a high temperature of 1185 °C. Molybdenum atoms underwent a self-assembly process driven by the electric field, followed by ultrafast cooling that solidified its intrinsic metallic properties, allowing the metastable structure to exist stably.
Breakthroughs in the field of solid-state electrolytes also highlight the unique value of this technology. The metastable halide argyrodite electrolyte (Li6PS5X, X = Cl−, Br−, I−) material constructed by the team of Annalise E. Maughan47 is based on the core mechanism of “transient ultra-high temperature – ultrafast cooling”: transient ultra-high temperatures of 800–1185 °C provided energy for lattice reconstruction, and ultrafast cooling froze the non-equilibrium lattice. In the Li6PS5I system, rapid quenching reduced the I− occupancy at the 4a/4d sites, breaking the ordered arrangement of the anion sublattice and inducing high-amplitude rotational displacement of the PS43− tetrahedra. This activated the Li+ hopping pathway between cages and modulated the entropy barrier, resulting in a tenfold increase in room-temperature ionic conductivity (Fig. 6i).
The above cases demonstrate the core value of solvent-free microwave synthesis technology: achieving precise control of crystalline phases in phosphide systems, breaking structural symmetry limitations in two-dimensional materials, and enhancing ionic conduction performance through lattice disordering in solid-state electrolytes, achieving breakthroughs in the time scale, structural control, and performance gain. In the future, combining in situ characterization techniques to analyze the formation kinetics of metastable phases and extending the approach to diverse carrier systems can provide theoretical support for the design and application of unconventional phase materials.
3.2.2.6 Amorphous catalysts. Solvent-free microwave synthesis technology, leveraging its unique “ultrafast heating/cooling – kinetic freezing” characteristic, provides an innovative pathway for the efficient preparation of amorphous/crystalline composite metastable materials. Through the synergy of microwave dielectric heating and the low heat capacity of the solvent-free system, this technology achieves the rapid nucleation and structural freezing of precursors in an extremely short time, effectively breaking through the performance limitations of traditional crystalline materials. It lays a solid foundation for constructing metastable systems with high surface energy and high activity.
The research team of David Mitlin achieved a significant breakthrough in the preparation of cobalt molybdate/carbon nanotube metastable materials.50 The team successfully constructed a sub-nanocomposite with a core–shell structure using the solvent-free microwave method (Fig. 6j). Its formation mechanism fully embodies the unique advantages of ultrafast kinetics. Microwave irradiation rapidly heated the precursor to 260 °C, triggering a solid-phase reaction that generated a sub-nanosheet structure. Subsequently, an instantaneous cooling process at a rate of hundreds of °C per minute created a powerful kinetic barrier, successfully “freezing” the non-equilibrium structure. This resulted in the formation of an amorphous shell layer approximately 12 nm thick on the material's surface while maintaining the structural integrity of the crystalline core.
In this synthesis process, the carbon nanotube carrier plays a dual key role: it acts both as an efficient microwave absorber enhancing the energy focusing effect and as a heterogeneous nucleation site guiding the directional growth of crystals, significantly optimizing the formation pathway of the metastable phase. Meanwhile, the solvent-free environment places the system in a highly supersaturated state, further promoting the synergistic progression of rapid nucleation and the amorphization process, ultimately constructing the unique crystalline–amorphous core–shell structure. This sophisticated structural design not only effectively addresses the technical bottlenecks of traditional hydrothermal methods but also, through the precise regulation of the amorphous/crystalline phase ratio and defect structure, endows the material with excellent energy storage performance.
4 Electrocatalysts and the energy sector
4.1 Hydrogen evolution reaction
As a pivotal technology within the hydrogen energy economy system, the efficient implementation of the HER critically relies on high-performance catalytic materials.73–79 Solvent-free microwave synthesis technology provides an innovative solution to overcome the activity and stability limitations of conventional catalysts through its rapid controllable heating characteristics and precise interfacial control capabilities. This section will systematically elaborate on the unique advantages and application breakthroughs of this technology in HER-catalytic systems.Building on efficient fabrication of fundamental catalytic systems, researchers have further explored structural designs to overcome noble metal loading limitations. For instance, our group reported ultrafine Ru–Mo2C@CNT catalysts synthesized via solvent-free microwave synthesis, demonstrating exceptional HER performance in alkaline media.4 Specifically, in 1.0 M KOH electrolyte, this catalyst achieves 10 mA cm−2 current density at merely 15 mV overpotential with a Tafel slope of 26 mV dec−1, significantly outperforming commercial Pt/C (Fig. 7a). It also delivers high current outputs at low overpotentials (56 mV@500 mA cm−2 and 78 mV@1000 mA cm−2) (Fig. 7b), while maintaining over 1000 h operational stability (Fig. 7c). These superior properties primarily originate from strong coupling interactions between Ru–Mo2C and the CNT carrier. Such interactions substantially enhance conductivity while increasing active site density and stability. Advanced structural analyses (XPS and TEM) confirm valence state modulation of Ru/Mo, heterojunction formation, and efficient interfacial charge transfer, showcasing the unique advantages of solvent-free microwave synthesis.
 | ||
| Fig. 7 (a) Comparison of overpotential changes at 10 mA cm−2 and exchange current density. (b) Polarization curves for Ru–Mo2C@CNT in 1.0 M KOH solution. (c) The current–time (i–t) curve of Ru–Mo2C@CNT under the temporal evolution of the potential required to maintain 500 mA cm−2 for 1000 h. Reproduced with permission.4 Copyright 2021, Nature Portfolio. | ||
With breakthroughs in efficiency and stability achieved by the Ru–Mo2C@CNT system, research focus has shifted toward overcoming noble metal loading limitations. Our research group developed an efficient and eco-friendly solvent-free microwave synthesis method through engineered anchoring sites and precise reaction time control, successfully preparing high-loading (47.1 wt%), highly stable, and uniformly dispersed Pt/KB-Oss-40 catalysts.3 This method achieves synthesis in just 40 s with a yield exceeding 93.1%, effectively overcoming metal agglomeration issues caused by prolonged heating in traditional thermal synthesis. Experimental results demonstrate outstanding acidic HER performance: Pt0.6/KB-Oss-40 exhibits a remarkable mass activity of 27.3 A mgpt−1 at 50 mV overpotential—over 6 times higher than that of commercial Pt/C (4.3 A mgpt−1) (Fig. 8a); its Tafel slope reaches 21.9 mV dec−1 (Fig. 8b), while significantly reduced charge transfer resistance indicates substantially enhanced electron transport efficiency (Fig. 8c). Notably, the catalyst maintains 98% initial activity after 10000 CV cycles and shows negligible decay during continuous polarization testing lasting five days. This exceptional stability primarily stems from strong coupling effects effectively suppressing Pt atomic migration and agglomeration. This strategy demonstrates excellent versatility, extendable to other noble metals like Pd and Ru, with further performance optimization achievable using diverse carbon supports (e.g., CNT and rGO).
 | ||
| Fig. 8 (a) The mass activity of Pt0.4/KB-OS5-40, Pt0.6/KB-OS5-40, Pt0.8/KB-OS5-40 and commercial Pt/C at an overpotential of 50 mV. (b) Tafel slope of Pt0.4/KB-OS5-40, Pt0.6/KB-OS5-40, Pt0.8/KB-OS5-40 and commercial Pt/C. (c) EIS Nyquist plots for Pt0.6/KB-OS5-40, Pd0.6/KB-OS5-40 and Ru0.6/KB-OS5-40. Reproduced with permission.3 Copyright 2022, RSC. (d) Comparison of overpotential at 10 mA cm−2. (e) Comparison of overpotential at 10 mA cm−2 in 1.0 mol L−1 KOH+ seawater. (f) Polarization curves for Ru2P@Ru/CNT, Ru2P/CNT, Ru/CNT and CC in 1.0 mol L−1 KOH solution. Reproduced with permission.22 Copyright 2022, Elsevier. | ||
Following significant achievements at laboratory current densities, catalyst adaptability in complex environments (such as seawater) becomes critical for industrial applications. Leveraging the instantaneous high-temperature reaction capability of solvent-free microwave technology, our group achieved rapid crystalline phase formation while effectively controlling nanoparticle size and minimizing volatile element loss. Taking the Ru2P@Ru/CNT heterostructure as an example: surfactant-free catalysts with ultra-small particle sizes (∼2.5 nm) were prepared within 1 minute using this method.22 The unique Ru/Ru2P multi-interface structure significantly optimizes hydrogen intermediate adsorption energetics through electronic coupling effects, enabling exceptional HER performance in both alkaline and seawater environments. Experimental data demonstrate that optimized Ru2P@Ru/CNT requires only 23 mV overpotential to reach 10 mA cm−2 in 1.0 M KOH, surpassing commercial Pt/C (33 mV) (Fig. 8d). In real seawater, it maintains a low overpotential of 29 mV (Fig. 8e), breaking the activity limitations of conventional catalysts in complex electrolyte systems. Notably, it achieves an overpotential of merely 104 mV at ultra-high current density (1000 mA cm−2) (Fig. 8f), with TOF values reaching 13.1 s−1 in freshwater and 8.5 s−1 in seawater, highlighting superior intrinsic activity. These outstanding properties stem from multiple advantages of microwave synthesis: rapid kinetics suppressing particle agglomeration, phosphide incorporation optimizing electronic structures, and CNT supports enhancing conductivity and overall stability. This green and efficient synthesis strategy provides innovative pathways for developing high-performance, non-noble metal seawater electrolysis catalysts, demonstrating broad application prospects in renewable hydrogen production.
Beyond noble metal systems, the microwave dielectric heating mechanism achieves efficient synthesis of non-noble metal nanocatalysts under solvent-free conditions. For instance, transition metals (e.g., Fe, Co, and Ni) and their complexes—as precursors for non-noble metal HER catalysts—can form highly dispersed nanostructures through microwave radiation-induced solid-state reactions. Xavier Bantreil's research team made significant advances in metal complex synthesis,80 efficiently preparing Group 8 and 9 metal complexes (such as [Fe(CO)3(L)] and [RuCl2(p-cymene)]2) via solvent-free microwave methods. These complexes were subsequently converted into metal/carbon composites through carbothermal reduction or in situ carbonization. Benefiting from rapid microwave heating, these materials exhibit high specific surface areas and uniform active site distribution, substantially enhancing their electrochemical activity. The team demonstrated that non-equilibrium thermal effects generated during microwave synthesis effectively inhibit particle agglomeration, yielding ultrafine (<5 nm) metal nanoparticles. This exposes more edge active sites and optimizes hydrogen adsorption free energy (ΔG*H). For example, cobalt complex-derived Co@N–C composites show HER activity approaching that of commercial Pt/C in alkaline media (η10 ≈ 52 mV at 10 mA cm−2). Compared to traditional solvothermal synthesis, solvent-free microwave methods not only reduce reaction times from h to mins but also enhance material durability and conductivity through precise control of crystalline phases and morphologies (e.g., constructing porous architectures or core–shell configurations).
Solvent-free microwave synthesis provides a straightforward, efficient, and environmentally friendly pathway for preparing high-performance electrocatalysts. By rationally designing the catalyst structure and composition, its electrocatalytic performance can be further optimized, offering new possibilities for industrial-scale hydrogen production.
4.2 Oxygen reduction reaction and complete water splitting
Building upon significant advances in high-efficiency HER catalysts, the research scope of solvent-free microwave synthesis technology has expanded to more complex energy conversion systems. As the core electrochemical process in fuel cells and metal–air batteries, the oxygen reduction reaction (ORR) directly determines the overall efficiency of related energy devices.23,81–87 Concurrently, overall water splitting technology—which efficiently integrates the HER with the OER88–93—stands out as a crucial pathway toward achieving carbon neutrality goals. Leveraging its precise interfacial control capabilities, solvent-free microwave synthesis technology opens up new avenues for constructing high-performance bifunctional catalysts.In the field of electrocatalysis, solvent-free microwave synthesis not only significantly enhances HER performance but also demonstrates marked advantages for ORR catalysis. For example, our group employed a microwave-assisted method to prepare PdRu/CNT catalysts that exhibit excellent performance in acidic ORR.1 Among them, the Pd69Ru31/CNT catalyst requires only 225 mV overpotential to achieve a current density of 10 mA cm−2 in 0.1 M HClO4 electrolyte (Fig. 9a). Its Tafel slope of 51.6 mV dec−1 far outperforms that of commercial Ir/C (Fig. 9b). Notably, the catalyst shows exceptional stability, with a mere ∼10 mV potential decay after 5000 CV cycles (Fig. 9c). Double-layer capacitance measurements (4.98 mF cm−2) indicate a higher electrochemically active surface area, surpassing that of commercial Ir/C by 61%. These improvements primarily stem from electronic structure optimization through bimetallic alloying, significantly enhancing the catalyst's adsorption and desorption capabilities toward oxygen intermediates.
 | ||
| Fig. 9 (a) OER polarization curves (scan rate: 5 mV s−1). (b) Histogram profile of overpotential at 10 mA cm−2 and Tafel slope. (c) OER polarization curves of Pd69Ru31/CNTs before and after 5000 testing cycles. Reproduced with permission.1 Copyright 2021, Wiley. (d) Comparison of over-potential changes at 10 mA cm−2. (e) Tafel plots obtained from the polarization curves. (f) The chronopotentiometric curves of the RuNiMoCrFeOx/CC and RuO2/CC electrodes tested at a constant current density of 100 mA cm−2. Reproduced with permission.20 Copyright 2022, RSC. | ||
Following breakthroughs in single-reaction catalytic performance, researchers have shifted focus toward integrated innovation of overall water splitting systems. Traditional setups typically require separate optimization of HER and OER catalysts, leading to complex device architectures and increased costs. Solvent-free microwave synthesis technology addresses this through multicomponent synergistic design, enabling the controlled preparation of efficient bifunctional catalysts—laying the material foundation for simplified electrolyzers. For instance, our group achieved rapid synthesis of RuNiMoCrFeOx/CNT high-entropy oxide catalysts within 120 s.20 This catalyst demonstrates breakthrough acidic OER performance: reaching 10 mA cm−2 at only 219 mV overpotential (significantly lower than commercial RuO2's 280 mV) (Fig. 9d) with an exceptional Tafel slope of 47 mV dec−1 (Fig. 9e). It maintains stable activity during 100 h continuous operation at 100 mA cm−2 (Fig. 9f). Isotope tracing and ESR analysis reveal that its catalytic mechanism primarily involves a lattice oxygen-mediated (LOM) pathway. Synergistic interactions among multimetallic elements effectively stabilize active sites, avoiding metal dissolution common in conventional adsorption-based mechanisms. The synthesis achieved gram-scale production (1.18 g per batch), yielding uniformly dispersed ∼6 nm nanoparticles on CNT supports—providing a scalable high-efficiency solution for proton exchange membrane (PEM) water electrolyzers.
Through its rapid and uniform heating characteristics, solvent-free microwave synthesis technology not only significantly shortens reaction times but also drastically reduces chemical waste generation by completely circumventing solvent usage. This technology demonstrates unique advantages in preparing high-performance ORR and overall water-splitting catalysts, offering new pathways for advancing the sustainable chemical industry.
4.3 Nitrogen reduction reaction
Against the backdrop of accelerating the global energy structure transition toward low-carbonization, green ammonia synthesis technologies are gaining widespread attention as crucial alternatives to the traditional Haber process. The nitrogen reduction reaction (NRR), with its capability to directly convert N2 to NH3 under mild conditions, is regarded as an ideal solution for distributed, low-energy ammonia production.94–99 However, conventional NRR catalysts universally face core challenges such as insufficient active sites, high nitrogen adsorption energy barriers, and interference from competitive HER, severely limiting their practical application efficiency. In this context, solvent-free microwave synthesis technology provides an innovative breakthrough pathway for developing high-performance NRR catalysts by leveraging its precise defect modulation capabilities and ultrafast reaction kinetics.Through a solvent-free microwave synthesis strategy, our research group successfully prepared ordered-vacancy sub-10 nm intermetallic PdM/CNT catalysts (M = Pb, Sn, In) within 60 s.5 This method achieves precise control of vacancy concentration by regulating microwave irradiation time (30–180 s), yielding catalyst production rates up to 82%. Among these, the OVs-Pd3Pb-2 catalyst optimized with 60 s irradiation demonstrates optimal vacancy distribution, achieving exceptional NRR performance at −0.05 V (vs. RHE): an NH3 yield rate of 88.3 µg h−1 mg−1, FE of 41.1% (Fig. 10a), and TOF of 27.3 h−1. Compared to vacancy-free Pd3Pb/CNT, it exhibits 7.7 times, 4.9 times, and 12.4 times enhancements in NH3 yield, FE, and TOF, respectively (Fig. 10b). DFT calculations reveal that Pd–Pb electronic synergy significantly strengthens nitrogen adsorption capacity, while the exposed Pd active sites at Pb vacancies reduce the reaction energy barrier for N2 → *NNH from 1.32 eV to 0.78 eV. This strategy displays excellent universality—extended to OVs–Pd2Sn and OVs–Pd3. In catalyst systems, it similarly demonstrates outstanding activity and stability, retaining 92% initial performance after 100 h of continuous operation (Fig. 10c).
 | ||
| Fig. 10 (a) Faradaic efficiencies (FEs) and corresponding NH3 yield of the intermetallic OVs-Pd3Pb-2 catalyst at various potentials in N2-saturated 0.1 M Li2SO4 electrolytes. (b) FE data and corresponding NH3 production of the intermetallic OVs-Pd3Pb-2 and comparison with different reported catalysts for the NRR. (c) Recycling measurement of the intermetallic OVs-Pd3Pb-2 at −0.05 V vs. RHE. Reproduced with permission.5 Copyright 2022, Elsevier. (d) NH3 formation rate over catalysts with different supports and promoters (533 K, 0.1 MPa). (e) Free energy (G-TS + ZPE) diagram of NH3 formation from the reaction of N2 and H2 over the Ru(211), K-promoted Ru(211) and Cs-promoted Ru(211) surfaces at 533 K. (f) NH3 production rate over the Ru(211), K–Ru(211) and Cs–Ru(211) surfaces in the temperature range of 500 K–700 K. Total pressure 1 bar, N2:H2 in a 1:3 ratio. Reproduced with permission.30 Copyright 2021, Elsevier. | ||
This technology also demonstrates significant application value in the field of ammonia synthesis catalyst development, offering new pathways for distributed ammonia production driven by renewable energy. Research by Jianli Hu's team revealed that in microwave-catalytic ammonia synthesis systems, Cs-promoted Ru/CeO2 catalysts exhibit breakthrough low-temperature activity.30 Under atmospheric pressure at 533 K, this catalyst achieves an ammonia production rate of 1.18 mmol h−1 gcat−1 (Fig. 10d), reducing energy consumption by 40% compared to conventional thermal catalytic processes. Its exceptional performance stems from a triple synergistic mechanism: the CeO2 carrier utilizes high dielectric loss properties and reversible Ce3+/Ce4+ redox couples to induce localized hotspots and generate Ov in microwave fields, significantly enhancing N2 adsorption and activation capabilities; Ru nanoparticles anchored via Ru–O–Ce bonds form highly dispersed active centers (average size: 6.1 nm), exposing abundant B5-type active sites; The Cs promoter transfers electrons to Ru due to its low electronegativity (χ = 0.79), causing a 0.9 eV negative shift in Ru 3d5/2 binding energy and reducing the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N dissociation energy barrier from 1.86 eV to 0.78 eV (DFT-verified) (Fig. 10e), thereby dramatically accelerating nitrogen hydrogenation kinetics (Fig. 10f).
N dissociation energy barrier from 1.86 eV to 0.78 eV (DFT-verified) (Fig. 10e), thereby dramatically accelerating nitrogen hydrogenation kinetics (Fig. 10f).
Despite facing numerous challenges in transitioning from laboratory research to industrial applications, solvent-free microwave synthesis technology demonstrates unique advantages that illuminate new directions for NRR catalyst design. Through its green and efficient preparation process, this method simultaneously optimizes active site density, electronic structure, and structural stability, successfully overcoming the technical limitations inherent in traditional synthesis approaches.
5 Challenges and prospects
The core value of strongly coupled sub-nanometer interface materials and metastable materials lies in establishing a completely new material design paradigm characterized by “atomic-level precise regulation – enhanced interface effects – non-equilibrium performance breakthrough,” bringing revolutionary progress to the field of energy catalysis (Fig. 11). Strongly coupled sub-nanometer interface materials, centered on active units smaller than 10 nanometers, address the key shortcomings of traditional nanocatalysts—such as the agglomeration of active sites and inefficient electron transport—through atomic/cluster-scale chemical bonding and electronic coupling. This achieves the synergistic optimization of atomic utilization, catalytic activity, and stability. Metastable materials, on the other hand, break the constraints of thermodynamic equilibrium by “freezing” high-energy metastable structures, such as those with high defect density, unconventional crystal phases, and atomic-level miscibility. This endows the materials with unique electronic configurations and reactivities that are unattainable by conventional stable phases. Both material types share the advantages of precise interface engineering, diversified structural design, and green synthesis pathways. They not only break through the bottlenecks in energy conversion efficiency but also propel catalytic science from empirical screening towards rational design, providing crucial support for the industrialization of efficient, low-cost energy technologies.100–102 | ||
| Fig. 11 Schematic summary of development logic for strongly coupled sub-nanoscale interface materials and metastable materials (note: dashed arrows indicate driving relationships; solid arrows indicate supporting relationships). | ||
The development of this field still faces dual challenges in both basic research and industrial application. At the basic research level, the understanding of strong coupling interactions is largely based on static characterization, lacking real-time tracking of interface chemical bond formation/breaking and electron transfer pathways during reactions. The correlation between the long-term evolution of defects in metastable materials and performance degradation remains unclear. Furthermore, a unified descriptor linking the interface microenvironment to catalytic performance has not been established, making it difficult to support the rational design of materials. For industrial application, metastable structures are prone to phase transformation or dissolution at high current density and in complex electrolyte environments. The high loading of precious metals increases material costs, while the performance of non-precious metal alternative systems still lags behind. Additionally, existing electrode materials face significant interfacial mass transfer resistance in membrane electrode assemblies, making it difficult to adapt to industrial equipment and hindering the technology's commercialization process.
Looking forward, the field requires dual-driver development through breakthroughs in basic research and technological application innovation. Basic research needs to utilize techniques such as in situ time-resolved spectroscopy and synchrotron radiation to analyze the dynamic interfacial coupling mechanisms. Combining multi-scale simulations and machine learning to build predictive models for metastable structure stability, and leveraging big data mining to construct quantitative structure–activity relationship systems are essential. Simultaneously, the functional boundaries of materials should be expanded into areas like coupled reactions and integrated photo/electro/thermal catalysis. On the technological application front, it is necessary to develop low-cost alternative systems based on non-precious metal high-entropy alloys or single-atom alloys, optimize solvent-free microwave synthesis equipment for continuous and scalable production, and tailor the hierarchical pore structures and interface compatibility of materials for specific energy devices, promoting the development of integrated electrode preparation technologies. Furthermore, enhancing industry-academia-research collaboration, focusing on pilot demonstrations in key scenarios like hydrogen production and green ammonia synthesis, will accelerate the translation of laboratory achievements into industrial products. This will fully unleash the core value of strongly coupled sub-nanometer interface materials and metastable materials in the low-carbon transition of the energy structure, contributing to the building of a sustainable energy system.
Author contributions
L. W. and J. L. supervised the review writing process. J. L. conceived the theme and framework of the review. Y. G. designed the review's structural content, collected most of the literature materials, and performed data analysis. W. X, H. L, and J. C provided technical expertise in relevant fields and assisted in revising the review. All authors participated in the review discussions, and examined and revised the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272222 and 52472217), Taishan Scholar Talent Program (tstp20250729), and University Youth Innovation Team of Shandong Province (202201010318).References
- Y. Qin, Z. Wang, Z. Fu, J. Lai and L. Wang, Electrochem. Sci. Adv., 2021, 2, e202100111 CrossRef.
- H. Miao, D. Zhang, Y. Shi, X. Wu, W. Zhang, X. Chen, J. Lai and L. Wang, ACS Sustainable Chem. Eng., 2021, 9, 15063–15071 Search PubMed.
- D. Wu, D. Zhang, Z. Wang, J. Xu, X. Chen, J. Lai and L. Wang, Mater. Chem. Front., 2022, 6, 3033–3041 RSC.
- X. Wu, Z. Wang, D. Zhang, Y. Qin, M. Wang, Y. Han, T. Zhan, B. Yang, S. Li, J. Lai and L. Wang, Nat. Commun., 2021, 12, 4810 CrossRef PubMed.
- Z. Wang, J. Liu, X. Wu, N. Nie, D. Zhang, H. Li, H. Zhao, J. Lai and L. Wang, Appl. Catal., B, 2022, 314, 121465 CrossRef CAS.
- C. Hu, R. Chen and N. Zheng, Adv. Mater., 2021, 33, 2006159 CrossRef CAS.
- X. Sun, C. Huang, L. Wang, L. Liang, Y. Cheng, W. Fei and Y. Li, Adv. Mater., 2021, 33, 2001105 CrossRef CAS PubMed.
- C. P. Theologides, G. G. Olympiou, P. G. Savva, K. Kapnisis, A. Anayiotos and C. N. Costa, Appl. Catal., B, 2017, 205, 443–454 CrossRef CAS.
- L. Liu and A. Corma, Nat. Catal., 2021, 4, 453–456 CrossRef CAS.
- M. Liu, Y. Xu, Y. Meng, L. Wang, H. Wang, Y. Huang, N. Onishi, L. Wang, Z. Fan and Y. Himeda, Adv. Energy Mater., 2022, 12, 2200817 CrossRef CAS.
- B. Zhao, W. Zeng, W. Zhang, S. Chen, H. Xu, Y. Liao, Y. Liao, Y. Qing and Y. Wu, Appl. Catal., B, 2024, 350, 123947 CrossRef CAS.
- B. Ni, Y. Shi and X. Wang, Adv. Mater., 2018, 30, 1802031 CrossRef PubMed.
- Y. Ding, X. Chen, Y. Zhou, X. Ren, W. Zhang, M. Li, Q. Zhang, T. Jiang, B. Ding, D. Shi and J. You, Adv. Sci., 2022, 9, 2201287 Search PubMed.
- Q. Lu and X. Wang, Adv. Sci., 2022, 9, 2104225 CrossRef CAS PubMed.
- T. Tsukamoto, K. Tomozawa, T. Moriai, N. Yoshida, T. Kambe and K. Yamamoto, Angew. Chem., Int. Ed., 2022, 61, e202201133 CrossRef.
- Z. Chen, Y. Zhang, C. Zhao, Y. Xia, Z. Li, X. Zhou, L. Xiao, X. Liu and Y. Zhang, Adv. Mater., 2024, 36, 2310022 Search PubMed.
- C. Feng, H. Wu, J. Shao, Q. Huo, A. Hassan, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2025, 15, 2403354 CrossRef CAS.
- X. Wang, Q. Liu and X. Wang, Adv. Funct. Mater., 2025, 35, 2504275 CrossRef CAS.
- H. Zhao, D. Zhang, Y. Yuan, X. Wu, S. Li, Z. Li, J. Lai and L. Wang, Appl. Catal., B, 2022, 304, 120916 CrossRef CAS.
- Y. Yu, H. Li, J. Liu, W. Xu, D. Zhang, J. Xiong, B. Li, A. O. Omelchuk, J. Lai and L. Wang, J. Mater. Chem. A, 2022, 10, 21260–21265 Search PubMed.
- S. Xu, G. Zhong, C. Chen, M. Zhou, D. J. Kline, R. J. Jacob, H. Xie, S. He, Z. Huang, J. Dai, A. H. Brozena, R. Shahbazian-Yassar, M. R. Zachariah, S. M. Anlage and L. Hu, Matter, 2019, 1, 759–769 CrossRef CAS.
- D. Zhang, H. Miao, X. Wu, Z. Wang, H. Zhao, Y. Shi, X. Chen, Z. Xiao, J. Lai and L. Wang, Chin. J. Catal., 2022, 43, 1148–1155 CrossRef CAS.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- D. Zhang, Y. Shi, H. Zhao, W. Qi, X. Chen, T. Zhan, S. Li, B. Yang, M. Sun, J. Lai, B. Huang and L. Wang, J. Mater. Chem. A, 2021, 9, 889–893 RSC.
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen, S. Li, J. Lai, B. Huang and L. Wang, Adv. Funct. Mater., 2021, 31, 2006939 Search PubMed.
- H. Li, M. Sun, Y. Pan, J. Xiong, H. Du, Y. Yu, S. Feng, Z. Li, J. Lai, B. Huang and L. Wang, Appl. Catal., B, 2022, 312, 121431 Search PubMed.
- D. Zhang, Y. Shi, X. Chen, J. Lai, B. Huang and L. Wang, Chin. J. Catal., 2023, 45, 174–183 CrossRef CAS.
- Y. Chen, Z. Lai, X. Zhang, Z. Fan, Q. He, C. Tan and H. Zhang, Nat. Rev. Chem., 2020, 4, 243–256 Search PubMed.
- S. Lin, T. Yu, Z. Yu, X. Hu and D. Yin, Adv. Mater., 2018, 30, 1705691 Search PubMed.
- Y. Wang, C. Wildfire, T. S. Khan, D. Shekhawat, J. Hu, P. Tavadze, R. Quiñones-Fernández and S. Moreno, Chem. Eng. J., 2021, 425, 139546 Search PubMed.
- B. Quan, A. Jin, S. H. Yu, S. M. Kang, J. Jeong, H. D. Abruña, L. Jin, Y. Piao and Y. E. Sung, Adv. Sci., 2018, 5, 1700880 CrossRef PubMed.
- Y. Zhang, H. Ren, T. Gao, J. Mou, Y. Meng, G. Tan, J. Ma and D. Xiao, Mater. Today Energy, 2020, 17, 100477 CrossRef.
- A. Taniguchi, R. Miyata, M. Ohtani and K. Kobiro, RSC Adv., 2022, 12, 22902–22910 RSC.
- P. Yang, F. Liu, X. Zang, L. Xin, W. Xiao, G. Xu, H. Li, Z. Li, T. Ma, J. Wang, Z. Wu and L. Wang, Adv. Energy Mater., 2023, 14, 2303384 CrossRef.
- B. Zhou, J. Wang, L. Guo, H. Li, W. Xiao, G. Xu, D. Chen, C. Li, Y. Du, H. Ding, Y. Zhang, Z. Wu and L. Wang, Adv. Energy Mater., 2024, 14, 2402372 CrossRef CAS.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 RSC.
- X. Sun, X. Wang, F. Sun, M. Tian, L. Qu, P. Perry, H. Owens and X. Liu, Adv. Mater., 2021, 33, 2105174 CrossRef CAS.
- J. Dickenson, Nat. Rev. Chem., 2024, 8, 873 CrossRef.
- D. J. Burke and D. J. Lipomi, Energy Environ. Sci., 2013, 6, 2053–2066 RSC.
- N. Nie, D. Zhang, Z. Wang, W. Yu, S. Ge, J. Xiong, Y. Gu, B. Yang, J. Lai and L. Wang, Appl. Catal., B, 2022, 318, 121808 CrossRef CAS.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- N. Nie, D. Zhang, Z. Wang, Y. Qin, X. Zhai, B. Yang, J. Lai and L. Wang, Small, 2021, 17, 2102879 CrossRef CAS.
- M. Wen, N. Sun, L. Jiao, S. Q. Zang and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202318338 CrossRef CAS PubMed.
- H. Fei, J. Dong, C. Wan, Z. Zhao, X. Xu, Z. Lin, Y. Wang, H. Liu, K. Zang, J. Luo, S. Zhao, W. Hu, W. Yan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2018, 30, 1802146 CrossRef.
- Y. Zhang, D. Zhang, Y. Qin, J. Xiong, J. Liu, W. Yu, X. Chen, S. Li, J. Lai and L. Wang, J. Energy Chem., 2022, 72, 108–115 CrossRef CAS.
- Z. Wu, Q. Li, G. Xu, W. Jin, W. Xiao, Z. Li, T. Ma, S. Feng and L. Wang, Adv. Mater., 2023, 36, 2311018 CrossRef PubMed.
- A. M. Shotwell, M. C. Schulze, P. Yox, C. Alaniz and A. E. Maughan, Adv. Funct. Mater., 2025, 35, 2500237 CrossRef CAS.
- X. Wu, W. Xu, Z. Wang, H. Li, M. Wang, D. Zhang, J. Lai and L. Wang, Chem. Eng. J., 2022, 431, 133247 CrossRef CAS.
- X. Fu, X. Zang, J. Gao, H. Li, W. Xiao, Y. Zong, G. Fu, J. Wang, T. Ma, W. Jin, Z. Wu and L. Wang, Adv. Energy Mater., 2025, 15, 2501054 CrossRef CAS.
- Z. Xu, Z. Li, X. Tan, C. M. B. Holt, L. Zhang, B. S. Amirkhiz and D. Mitlin, RSC Adv., 2012, 2, 2753 RSC.
- D. Wu, H. Du, Z. Liu, G. A. Bagliu, J. Lai and L. Wang, EcoEnergy, 2024, 2, 724–735 CrossRef CAS.
- J. Liu, Z. Wang, X. Wu, D. Zhang, Y. Zhang, J. Xiong, Z. Wu, J. Lai and L. Wang, J. Mater. Chem. A, 2022, 10, 15395–15401 RSC.
- Y. Shi, D. Zhang, H. Miao, W. Zhang, X. Wu, Z. Wang, H. Li, T. Zhan, X. Chen, J. Lai and L. Wang, J. Mater. Chem. A, 2021, 9, 24261–24267 RSC.
- F.-Z. Zhang, X.-B. Liu, C.-M. Yang, G.-D. Chen, Y. Meng, H.-B. Zhou and S.-H. Zhang, Mater. Today, 2024, 74, 203–234 CrossRef CAS.
- A. Tundwal, H. Kumar, B. J. Binoj, R. Sharma, R. Kumari, A. Yadav, G. Kumar, A. Dhayal, A. Yadav, D. Singh, B. Mangla and P. Kumar, Coord. Chem. Rev., 2024, 500, 215533 CrossRef CAS.
- P. Xie, W. Yuan, X. Liu, Y. Peng, Y. Yin, Y. Li and Z. Wu, Energy Storage Mater., 2021, 36, 56–76 CrossRef.
- A. R. Deline, B. P. Frank, C. L. Smith, L. R. Sigmon, A. N. Wallace, M. J. Gallagher, D. G. Goodwin, Jr., D. P. Durkin and D. H. Fairbrother, Chem. Rev., 2020, 120, 11651–11697 Search PubMed.
- Y. Tanabe, Y. Ito, K. Sugawara, S. Jeong, T. Ohto, T. Nishiuchi, N. Kawada, S. Kimura, C. F. Aleman, T. Takahashi, M. Kotani and M. Chen, Adv. Mater., 2022, 34, 2205986 Search PubMed.
- Z. Li, Y. Chen, T. Ma, Y. Jiang, J. Chen, H. Pan and W. Sun, Adv. Energy Mater., 2021, 11, 2101202 Search PubMed.
- B. Gao, M. Sun, W. Ding, Z. Ding and W. Liu, Appl. Catal., B, 2021, 281, 119492 Search PubMed.
- J. Chen, H. Li, C. Fan, Q. Meng, Y. Tang, X. Qiu, G. Fu and T. Ma, Adv. Mater., 2020, 32, 2003134 CrossRef CAS PubMed.
- Y. Zhang, G. Hai, Z. Huang, Z. Liu, X. Huang and G. Wang, Adv. Energy Mater., 2024, 14, 2401449 CrossRef CAS.
- Y.-Y. Wang, Z.-Y. Wang, Y.-J. Xu, W.-H. Chen, G.-S. Shao and B.-H. Hou, Energy Environ. Sci., 2024, 17, 6811–6820 Search PubMed.
- M. You, X. Du, X. Hou, Z. Wang, Y. Zhou, H. Ji, L. Zhang, Z. Zhang, S. Yi and D. Chen, Appl. Catal., B, 2022, 317, 121729 Search PubMed.
- H. Jia, M. Qiu, C. Tang, H. Liu, J. Xu, B. Tawiah, S.-x. Jiang and X. Zhang, Adv. Fiber Mater., 2022, 4, 1500–1510 Search PubMed.
- T. Zhang, W. Zong, Y. Ouyang, Y. Wu, Y.-E. Miao and T. Liu, Adv. Fiber Mater., 2021, 3, 229–238 CrossRef CAS.
- D. Zhang, Z. Wang, X. Wu, Y. Shi, N. Nie, H. Zhao, H. Miao, X. Chen, S. Li, J. Lai and L. Wang, Small, 2021, 18, 2104559 Search PubMed.
- J. Liu, Z. Wang, D. Zhang, Y. Qin, J. Xiong, J. Lai and L. Wang, Small, 2022, 18, 2108072 CrossRef CAS.
- Y. Lin, D. W. Baggett, J.-W. Kim, E. J. Siochi and J. W. Connell, ACS Appl. Mater. Interfaces, 2011, 3, 1652–1664 Search PubMed.
- X. Luan, L. Guo, P. Yang, W. Xiao, H. Li, L. Xin, G. Xu, T. Ma, D. Chen, Z. Wu and L. Wang, Fuel, 2024, 378, 132814 CrossRef CAS.
- Y. Shen, W. Li, W. Wang, L. Xin, W. Xiao, G. Xu, D. Chen, L. Wang, F. Liu and Z. Wu, Inorg. Chem. Front., 2024, 11, 5508–5516 RSC.
- T. K. Sahu, N. Kumar, S. Chahal, R. Jana, S. Paul, M. Mukherjee, A. H. Tavabi, A. Datta, R. E. Dunin-Borkowski, I. Valov, A. Nayak and P. Kumar, Nat. Nanotechnol., 2023, 18, 1430–1438 CrossRef CAS.
- Z. Wang, X. Zhang, X. Wu, Y. Pan, H. Li, Y. Han, G. Xu, J. Chi, J. Lai and L. Wang, Chem. Eng. J., 2022, 437, 135375 CrossRef CAS.
- J. Chi, L. Guo, J. Mao, T. Cui, J. Zhu, Y. Xia, J. Lai and L. Wang, Adv. Funct. Mater., 2023, 33, 2300625 CrossRef CAS.
- P. Liu, Y. Shi, X. Zhang, J. Yin, D. Zhang, T. Wang, J. Fei, T. Zhan, G. Li, J. Lai and L. Wang, Appl. Catal., B, 2024, 341, 123332 CrossRef CAS.
- P. Liu, X. Zhang, J. Fei, Y. Shi, J. Zhu, D. Zhang, L. Zhao, L. Wang and J. Lai, Adv. Mater., 2024, 36, 2310591 CrossRef CAS PubMed.
- X. Liu, X. Wang, K. Li, J. Tang, J. Zhu, J. Chi, J. Lai and L. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316319 CrossRef CAS.
- Y. Shi, J. Fei, H. Li, C. Li, T. Zhan, J. Lai and L. Wang, Appl. Catal., B, 2024, 359, 124465 CrossRef CAS.
- W. Xu, D. Zhang, T. Wang, J. Lai and L. Wang, Appl. Catal., B, 2025, 361, 124626 CrossRef CAS.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS.
- J. Lai and S. Guo, Small, 2017, 13, 1702156 CrossRef.
- J. Lai, B. Huang, Y. Tang, F. Lin, P. Zhou, X. Chen, Y. Sun, F. Lv and S. Guo, Chem, 2018, 4, 1153–1166 Search PubMed.
- J. Lai, F. Lin, Y. Tang, P. Zhou, Y. Chao, Y. Zhang and S. Guo, Adv. Energy Mater., 2019, 9, 1800684 CrossRef.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS.
- S. Wu, X. Liu, H. Mao, T. Cui, B. Li, G. Zhou and L. Wang, Appl. Catal., B, 2023, 330, 122634 CrossRef CAS.
- L. Zong, K. Fan, P. Li, F. Lu, B. Li and L. Wang, Adv. Energy Mater., 2023, 13, 2203611 CrossRef CAS.
- S. Wu, X. Liu, H. Mao, J. Zhu, G. Zhou, J. Chi, Z. Wu and L. Wang, Adv. Energy Mater., 2024, 14, 2400183 CrossRef CAS.
- I. Vinogradov, S. Singh, H. Lyle, M. Paolino, A. Mandal, J. Rossmeisl and T. Cuk, Nat. Mater., 2022, 21, 88–94 CrossRef CAS.
- B. Deng, G. Yu, W. Zhao, Y. Long, C. Yang, P. Du, X. He, Z. Zhang, K. Huang, X. Li and H. Wu, Energy Environ. Sci., 2023, 16, 5210–5219 RSC.
- B. Guo, Y. Ding, H. Huo, X. Wen, X. Ren, P. Xu and S. Li, Nano-Micro Lett., 2023, 15, 57 CrossRef CAS.
- H. S. Lee, H. Shin, S. Park, J. Kim, E. Jung, W. Hwang, B.-H. Lee, J. M. Yoo, W. H. Antink, K. Lee, S. Lee, G. Na, K. Suh, Y. S. Kim, K.-S. Lee, S. J. Yoo, Y.-E. Sung and T. Hyeon, Joule, 2023, 7, 1902–1919 CrossRef CAS.
- L. Magnier, G. Cossard, V. Martin, C. Pascal, V. Roche, E. Sibert, I. Shchedrina, R. Bousquet, V. Parry and M. Chatenet, Nat. Mater., 2024, 23, 252–261 CrossRef CAS.
- W. Feng, G. Zou, T. Liu, R. Li, J. Yu, Y. Guo, Q. Liu, X. Zhang, J. Wang, N. Ta, M. Li, P. Zhang, X. Cao, R. Yu, Y. Song, M. Liu, G. Wang and X. Bao, Energy Environ. Sci., 2025, 18, 2273–2284 RSC.
- J. Choi, H.-L. Du, C. K. Nguyen, B. H. R. Suryanto, A. N. Simonov and D. R. MacFarlane, ACS Energy Lett., 2020, 5, 2095–2097 CrossRef CAS.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. B. V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS.
- M. P. Maman, T. Gurusamy, A. K. Pal, R. Jana, K. Ramanujam, A. Datta and S. Mandal, Angew. Chem., Int. Ed., 2023, 62, e202305462 CrossRef CAS.
- A. Bagger, R. Tort, M.-M. Titirici, A. Walsh and I. E. L. Stephens, ACS Energy Lett., 2024, 9, 4947–4952 CrossRef CAS PubMed.
- S. Yang, J. Chu, J. Park, H. Kim and B. Shin, Angew. Chem., Int. Ed., 2024, 63, e202411909 CrossRef CAS.
- Y. Feng, L. Jiao, X. Zhuang, Y. Wang and J. Yao, Adv. Mater., 2025, 37, 2410909 CrossRef CAS.
- C. G. Lin, W. Zhou, X. T. Xiong, W. Xuan, P. J. Kitson, D. L. Long, W. Chen, Y. F. Song and L. Cronin, Angew. Chem., Int. Ed., 2018, 57, 16716–16720 CrossRef CAS PubMed.
- O. C. Pore, A. V. Fulari, R. V. Shejwal, V. J. Fulari and G. M. Lohar, Chem. Eng. J., 2021, 426, 131544 Search PubMed.
- S. Hong, S. Jin, Y. Deng, R. Garcia-Mendez, K.-i. Kim, N. Utomo and L. A. Archer, ACS Energy Lett., 2023, 8, 1744–1751 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |