
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoll-to-roll fabrication of integrated cathodes enabled by asymmetric dual-atom catalysts for bipolar stacking ampere-hour-scale Zn–air batteries
Yuanhao
Wei
a,
Yuanjie
Ma
b,
Jingjing
Huang
a,
Chenbao
Lu
 c,
Longbin
Li
d,
Xuejiao J.
Gao
*b,
Dirk
Lützenkirchen-Hecht
e,
Xiaodong
Zhuang
c,
Kai
Yuan
*a and
Yiwang
Chen
*ad
c,
Longbin
Li
d,
Xuejiao J.
Gao
*b,
Dirk
Lützenkirchen-Hecht
e,
Xiaodong
Zhuang
c,
Kai
Yuan
*a and
Yiwang
Chen
*ad
aCollege of Chemistry and Chemical Engineering, Institute of Polymers and Energy Chemistry (IPEC), Film Energy Chemistry for Jiangxi Provincial Key Laboratory (FEC), Nanchang University, 999 Xuefu Avenue, Nanchang 330031, China. E-mail: kai.yuan@ncu.edu.cn; ywchen@ncu.edu.cn
bCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China. E-mail: gaoxj@jxnu.edu.cn
cThe Soft2D Lab, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Dongchuan Road 800, Shanghai 200240, China
dCollege of Chemistry and Chemical Engineering, Gannan Normal University, Ganzhou 341000, China
eFaculty of Mathematics and Natural Sciences-Physics Department, Bergische Universität Wuppertal, Gauss-Str. 20, D-42119 Wuppertal, Germany
First published on 9th December 2025
Abstract
Zn–air batteries (ZABs) represent a highly promising electrochemical energy storage technology, yet their practical deployment remains constrained by several fundamental challenges, including sluggish oxygen reaction kinetics at the cathode, limited power output and capacity in conventional device architectures. Herein, we demonstrate an efficiently integrated strategy to systematically address these issues. First, a metal–semimetal dual-atom catalyst consisting of Fe–Se dual-atom sites (FeSe-NC) is synthesized, whose unique electronic structure and metal–semimetal synergistic effects significantly enhanced oxygen electrocatalytic activity, achieving a half-wave potential of 0.920 V and a turnover frequency of 0.77 e− site−1 s−1. Furthermore, a scalable roll-to-roll process was developed to fabricate an integrated air cathode with a highly uniform catalytic layer structure. Ultimately, the electrode was employed in a bipolar stacking cell configuration. Benefiting from these multi-level innovations, the assembled ampere-hour-scale ZABs delivered a high power output of 3.5 W, a large capacity of 6.09 Ah, and long-term cycling stability over 60 h at 1.0 A. This work systematically advances the development of high-performance, long-lasting ZABs through catalyst design, electrode engineering, and device configuration optimization, providing a viable technical pathway for their industrialization.
Introduction
The urgent need to combat climate change and address global energy shortages is accelerating demand for renewable energy sources. Owing to their high theoretical energy density, low cost, environmental friendliness, and excellent safety, rechargeable Zn–air batteries (ZABs) have emerged as a leading candidate for next-generation energy storage and conversion systems.1–3 The oxygen reduction reaction (ORR) serves as the critical cathodic process governing the energy conversion efficiency in ZABs,4,5 but the intrinsically sluggish kinetics of the ORR process severely restricts the performance and the practical application of these devices.6,7 In recent years, although developing advanced ORR catalysts remains a primary strategy for enhancing the performance of ZABs, diminishing returns in performance improvement indicate an emerging research bottleneck. This stagnation stems from disproportionate focus on catalytic activity at the expense of system-level implementation factors, such as tunable cell architectures and electrode fabrication techniques. Therefore, to achieve high-energy ZABs, it is imperative to strategically integrate multiscale coordination among catalyst activity optimization, electrode structural engineering, and battery system architecture, thereby establishing a trinity-coupled synergy framework for performance enhancement.Tracing the development of ORR electrocatalysts, single-atom electrocatalysts (SACs) have attracted great concerns for oxygen electrocatalysis because of their high atom utilization and favorable intrinsic activities.8–10 Nevertheless, symmetric charge configuration and inherent linear scaling relationships in SACs impose intrinsic limitations on catalytic activity advancement.11–15 The presence of adjacent metal pairs or long-range coupling effects in dual-atom catalysts (DACs) provides additional avenues for precisely regulating intermediate adsorption. This facilitates enhanced reaction kinetics and catalytic activity while overcoming the theoretical limits of the SACs.16–19 However, the current studies of DACs mainly focused on the 3d-block transition metal pairs.20–22 Due to the similar electronic structures and atomic dimensions of the 3d-block atom pairs, these catalysts exhibit limited efficiency in breaking symmetric charge distribution.23 Moreover, achieving precise and controllable synthesis of dual-atom catalysts still presents a significant obstacle. Therefore, precisely constructing dual-atom catalysts and designing more asymmetric charge distributions within them to enhance their performance has become a critical issue that urgently needs to be addressed.
Achieving high-performance ZABs requires not only the optimization of the intrinsic activity of electrocatalysts but also critically depends on the effective integration of these catalysts within the air cathode architecture. As shown in Fig. 1a, the production of conventional electrodes centers on a wet spray-coating process involving slurry preparation and drying. However, the inherent challenges of rheological control during slurry mixing and the complex multiphase behavior during drying hinder the formation of an ideal microstructure.24 This will trigger a series of adverse consequences, such as non-uniform coating layers and increased contact resistance due to excessive particle gaps.25 Furthermore, traditional electrodes based on carbon paper or carbon cloth substrates are severely constrained in their high-current-discharge performance. Besides, the inherently complex manufacturing processes of this method are also not conducive to scalable production.26 A further major challenge in designing high-energy ZABs stems from the battery architecture. The inhomogeneous surfaces common in traditional batteries inevitably trigger side reactions, leading to severe Zn loss that ultimately diminishes Zn utilization efficiency and compromises cycling lifespan (Fig. 1b).27 Ultimately, the cumulative effects of these defects severely constrain power output, rendering such designs unsuitable for high-performance applications. Thus, it remains a central challenge for ZABs to firmly integrate advanced catalysts into air cathodes within a rationally designed battery structure while enabling scalable manufacturing and retaining structural stability and electrochemical performance under high-current-density conditions.28
 | ||
| Fig. 1 Schematic diagram comparing (a) roll-to-roll vs. wet spray-coating processes and (b) conventional vs. bipolar stacking ZAB structures. | ||
To address these challenges, the key lies in the synergistic integration of catalyst design, air cathode engineering, and battery configuration. First, the combination of metal–semimetal is an effective approach to enhance more asymmetric electron distribution and further increase the catalytic activity of DACs. As a semi-metallic element, the selenium atom (Se) has unique physical and chemical properties, such as strong polarizability and abundant d electrons.29–31 These features may enable push–pull electronic effects between selenium and 3d-block transition metals with unpaired electrons, thereby disturbing their symmetric electronic structure distribution.32 With this measure, the adsorption energy barriers of the oxygen intermediates can be effectively regulated, thus decreasing the overpotential of the reaction.
However, conventional synthesis suffers from poorly controlled atomic arrangements during the high-temperature process, leading to ill-defined active sites and poor reproducibility. By employing a pre-polymerization strategy to build a structurally pre-defined precursor, well-defined dual-atom sites can be deterministically constructed. This precise methodology ensures exceptional control over the atomic coordination and, crucially, enables the scalable production of the catalyst with consistent structural fidelity and catalytic performance. Second, achieving superior electrochemical performance in ZABs requires efficient ion/electron transport pathways, which depend on the uniform distribution of electrode components. This goal can be effectively addressed by the roll-to-roll process, which offers the inherent advantage of fabricating dense air electrodes with highly homogeneous microstructures and binder distribution (Fig. 1a).33 The resulting structural uniformity promotes effective mass transfer, thereby enhancing performance.34 Coupled with significantly reduced manufacturing time and cost, these synergistic benefits critically advance ZAB commercialization.35 Third, unlike the conventional battery configuration, by balancing interfacial charges, the bipolar stacking cell structure with a doubled active area effectively curbs side reactions and reduces overpotential while boosting Zn utilization efficiency. These synergistic effects collectively enhance the battery's energy storage capacity, power output, and cycle life.27
Herein, we achieve a systematic performance enhancement in ZABs through the synergistic integration of catalyst construction, electrode structure engineering, and battery configuration innovation. First, a synchronous complexation-polymerization strategy to precisely synthesize DACs with asymmetric Fe–Se dual-atom sites (FeSe-NC) was developed. The polymerization of pyrrole and selenophenol, induced by FeCl3, creates a well-defined precursor with pre-organized Fe and Se atoms, which upon carbonization, guarantees the formation of the Fe–Se dual-atom structure with exceptional batch-to-batch consistency. Scanning electrochemical microscopy quantitatively verified that FeSe-NC attains an active site density of 6.40 × 1020 sites g−1, coupled with a high turnover frequency of 0.77 e− site−1 s−1. Theoretical calculations justify that the combination of Fe–Se dual-atom sites break the symmetric charge distribution on the Fe sites while modulating the binding strength of the oxygenated species, thus yielding exceptional ORR activity.
Consequently, FeSe-NC showcases remarkable ORR performance with a half-wave potential of 0.920 V under alkaline conditions. Subsequently, leveraging the as-developed catalyst, an integrated air cathode was fabricated via a scalable roll-to-roll process, enabling large-area electrode production. Benefiting from the inherent advantages of this technique, the resulting cathode exhibits a uniformly distributed catalytic layer. Furthermore, the repeated rolling process yields a densely packed architecture that significantly reduces interfacial contact resistance. Finally, air cathodes with different catalyst loadings were assembled into both conventional and bipolar stacking ZAB configurations. The bipolar stacking cell configuration achieves a remarkable power output of 3.5 W, an ultrahigh capacity of 6.09 Ah, and a cycle life exceeding 60 h at 1.0 A, significantly outperforming its conventional counterpart. These findings confirm that the integrated strategy combining catalyst design, electrode fabrication, and battery architecture optimization provides an effective and viable approach for developing high-performance ZABs.
Results and discussion
Synthesis and characterization of the catalyst
The synthesis of Fe–Se dual-atom sites anchored on porous nitrogen-doped carbon (FeSe-NC) is schematically illustrated in Fig. 2a. First, FeCl3 was used to initiate the polymerization of selenophenol and pyrrole to generate iron-containing pyrrole–selenophenol polymer hydrogel precursors. Afterward, the hydrogel was calcined in a tubular furnace at 1000 °C, and the FeSe-NC sample was obtained after leaching with H2SO4 solution. For comparison, FeNC (Se-free), SeNC (Fe-free), and FeSe-NC-X (X represents the calcination temperatures, X = 700, 800, 900, 1050) catalysts were prepared using a similar recipe.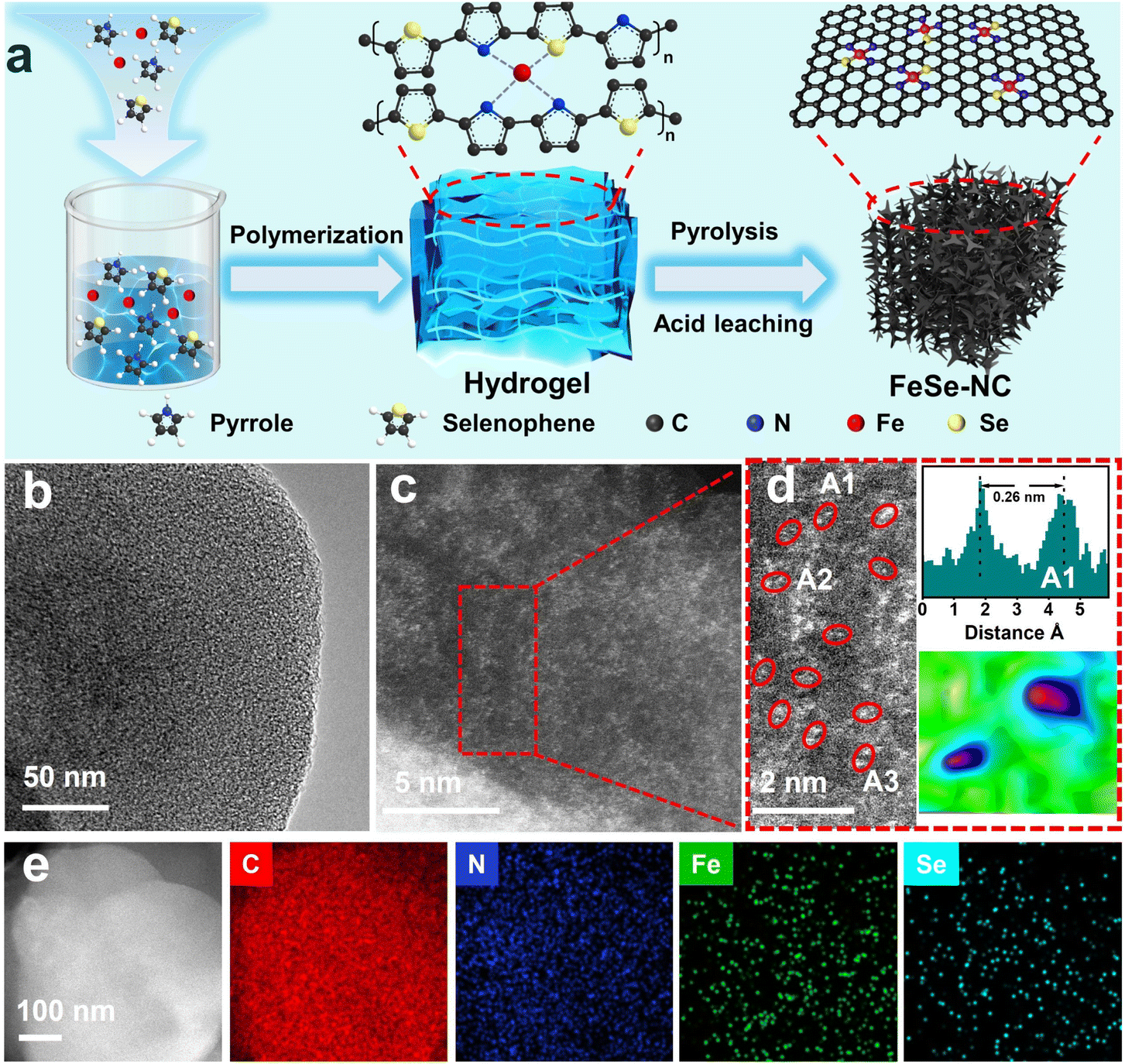 | ||
| Fig. 2 (a) Schematic illustration of the synthesis procedure of FeSe-NC. (b) TEM image.(c) Aberration-corrected HAADF-STEM image, (d) the left panel shows the enlarged image in the red box in (c), and the right panel displays the intensity distribution and the corresponding intensity profile in the A1 region. (e) HAADF-STEM image and corresponding elemental mapping of C, N, Fe and Se. | ||
The micro-morphology of the obtained catalysts was initially observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The obtained FeSe-NC possesses a morphology composed of interconnected coral-like nanofibers (Fig. S1, SI). It is also seen that FeNC exhibits the same coral-like nanofibers, while SeNC and NC show a clear bulk morphology (Fig. S2–4, SI). It is worth remarking that no obvious Fe and Se related nanoparticles (NPs) could be detected in the corresponding TEM images (Fig. 2b). The aberration-corrected high-angle annular dark field-scanning transmission electron microscopy (AC HAADF-STEM) images (Fig. 2c–d) further elucidated the distribution of Se and Fe species with obvious dual-atom sites (marked with red circles). Z contrast analysis serves as a powerful tool for qualitatively identifying the existence of heteronuclear atom pairs.36 Because of the different Z contrasts of Fe and Se, the presence of Fe–Se dual-atom sites in region A1 was confirmed by two bright spots of different intensities (Fig. 2d). Parallel analyses of other zones (Fig. S5, SI) indicate the prevalence of Fe–Se atomic pairs. The distance between the two heteronuclear atoms was further measured to be about 0.26 nm on average (Fig. 1d and S5, SI). The energy dispersion spectroscopy (EDS) mapping indicates a uniform distribution of Fe, Se, and N elements within the FeSe-NC catalyst (Fig. 2e).
X-ray diffraction (XRD) patterns of the obtained catalysts exhibit two characteristic diffraction peaks at 24° and 44°, which correspond to the (002) and (101) planes of carbon,37 respectively, indicating the absence of Fe and Se related NPs (Fig. S6, SI). The diffraction peaks of FeSe-NC are relatively weaker and wider compared to FeNC, demonstrating that Se doping leads to a higher density of defects. The Raman spectra (Fig. S7, SI) reveal that FeSe-NC exhibits a higher D-band to G-band intensity ratio (ID/IG = 1.13) compared to FeNC (0.89), further implying the presence of more defects induced by the introduction of Se.31 The porous structure and Brunauer–Emmett–Teller (BET) specific surface area were further investigated by N2 adsorption–desorption isotherms (Fig. S8 and Table S1, SI). The pore size distribution reveals that microporous and mesoporous structures co-exist in FeSe-NC. Moreover, FeSe-NC demonstrates a higher BET specific surface area than FeNC, thereby enhancing the accessibility of active sites in catalytic processes.
The chemical states of elements in the obtained catalysts were identified by X-ray photoelectron spectroscopy (XPS). As revealed by the survey spectra (Fig. S9 and Table S2, SI), Fe, Se, N, and C elements can be found in FeSe-NC. The high-resolution C 1s spectra reveal the formation of C–N (285.5 eV) and C–Se (289.9 eV) bonds,38 demonstrating the successful doping of N and Se (Fig. S10, SI). The high-resolution N 1s spectra of FeSe-NC are each resolved into five sub-peaks (Fig. S11, SI), corresponding to pyridinic N (398.4 eV), Fe–N (399.6 eV), pyrrolic N (400.7 eV), graphitic N (401.4 eV), and oxidized N (403.9 eV), respectively.39 With increasing pyrolysis temperature, the amounts of Fe–N and pyridinic N decrease, whereas the content of graphitic N increases (Fig. S12 and Table S3, SI). The Se 3d XPS spectrum of FeSe-NC exhibits three peaks centered at 59.0, 56.4, and 54.0 eV, corresponding to Se-Ox, Se–C, and Se–Fe bonds (Fig. S13, SI),31,40 verifying the formation of Se–Fe coordination. Compared to SeNC, the Se 3d peak of FeSe-NC is found to be positively shifted by 0.5 eV, which indicates that there is an electron transfer taking place between Fe and Se. The deconvolution of the high-resolution Fe 2p state of FeSe-NC reveals three distinct groups of peaks (Fig. 3a). The doublet at 708.3 and 711.0 eV is attributed to Fe 2p3/2, and the doublet at 721.7 and 724.4 eV is ascribed to Fe 2p1/2. The other doublet located at 715.3 and 728.8 eV is associated with satellite peaks.41 The Fe 2p binding energy of FeSe-NC shows a negative shift of 0.2 eV compared to FeNC. The results indicate that Se-modified Fe exhibits a lower valence state, which is attributed to the electron donating effect of the Se atom.
 | ||
| Fig. 3 (a) XPS profiles of Fe 2p spectra for FeSe-NC and FeNC. (b) XANES spectra at the Fe K-edge of FeSe-NC, FeNC, Fe2O3, FePc, Fe foil, and FeO. (c) Linear fitting curve of FeSe-NC, FeNC, and reference materials derived from the corresponding Fe K-edge XANES spectra. (d) The corresponding Fourier-transformed Fe K-edge EXAFS spectra. (e) WT-EXAFS for the Fe element of FeSe-NC, FeNC, FePc, and Fe foil. (f) The corresponding Fourier-transformed Se K-edge EXAFS spectra. (g) The corresponding fitting results of FeSe-NC in R space. The inset illustrates the proposed Fe center coordination configuration. | ||
The coordination information and atomic structure of catalysts were gained by further analyzing the K-edge XANES (X-ray absorption near-edge structure) and EXAFS (extended X-ray absorption fine structure) characterization studies (Fig. 3b). The first-order derivatives of the Fe K-edge XANES spectra (Fig. 3c and S14, SI) were utilized to further reveal the valence states of Fe. The linear fitting analysis indicates that the oxidation state of Fe in FeSe-NC is around +1.4, which is slightly lower than that of Fe in FeNC (+1.6). This demonstrates that the incorporation of Se effectively decreased the oxidation state of Fe, aligning well with the findings from the XPS analysis. In the Fourier transformed (FT) EXAFS spectra of Fe (Fig. 3d), one dominant peak (1.5 Å) and one shoulder peak (1.90 Å) attributed to Fe–N and Fe–Se coordination scattering from the first shell layer, respectively, can be detected. Fe–Fe bond-associated peaks are notably absent, which verifies that Fe in FeSe-NC is chiefly present as isolated single atoms. Wavelet transform EXAFS (WT-EXAFS) of FeSe-NC exhibits plots similar to those of FePc, distinctly different from Fe foil, further validating the atomic-scale dispersion of Fe within FeSe-NC (Fig. 3e). Similarly, the XANES (Fig. S15, SI) and EXAFS (Fig. 3f) spectra at the Se K-edge for FeSe-NC provide evidence for the existence of the Se–Fe bond. This further implies the formation of an Fe–Se dual atom configuration.42 Moreover, the least-squares EXAFS fitting analysis shows that the coordination numbers of Fe–N, Fe–Se, and Fe–C in FeSe-NC are 2.8, 1.1, and 4.2, respectively (Fig. 3g and Table S4, SI). This indicates that the Fe center in FeSe-NC predominantly adopts an FeN3Se1 configuration (inset in Fig. 3g), coordinated by three N atoms and one Se atom.
Electrocatalytic performance
The ORR electrocatalytic activity of the catalysts was evaluated using a rotating disk electrode (RDE) in an O2-saturated 0.1 M KOH electrolyte. The linear sweep voltammetry (LSV) curves (Fig. 4a and S16, SI) indicate that FeSe-NC shows the best ORR performance with a half-wave potential (E1/2) of 0.920 V vs. RHE, higher than most of the recently reported ORR catalysts (Tables S5 and 6, SI). To reveal the reaction kinetics, LSV curves were collected at different rotational speeds (Fig. S17 and 18, SI). The Tafel slope is 56.2 mV dec−1 for FeSe-NC (Fig. 4b and S19, SI), which is considerably lower than those for FeNC (87.9 mV dec−1) and Pt/C (84.6 mV dec−1). Meanwhile, FeSe-NC reaches a kinetic current density (jk) of 40.21 mA cm−2 at 0.85 V (Fig. 4c and S20, SI), which is nearly 3.5 and 4.4 times higher than that of FeNC (11.35 mA cm−2) and Pt/C (9.20 mA cm−2), respectively, suggesting the fast ORR kinetics of FeSe-NC. The number of electrons transferred during the ORR process was calculated from the Koutecký–Levich plot as 3.95 (Fig. S17, SI), implying FeSe-NC undergoes a direct four-electron transfer process. The ring-disk electrode (RRDE) test further validated the result (Fig. 4d and S21, SI). Within the potential range from 0.2 to 0.8 V, the H2O2 yield of FeSe-NC remains constantly below 6.22%, which is less than that of FeNC (11.41%). These findings confirm that the incorporation of Se enhances the four-electron selectivity. By comparing the Eonset, E1/2, jk, jL, and Tafel slope, it was found that FeSe-NC is a superior ORR catalyst with high catalytic activity and fast reaction kinetics (Fig. 4e). | ||
| Fig. 4 Electrocatalytic ORR performance of FeSe-NC. (a) LSV polarization curves, (b) Tafel plots, and (c) E1/2 and jk of FeSe-NC, FeNC, SeNC, NC, and Pt/C-20%. (d) H2O2 yield (%) and electron transfer number (n) obtained from RRDE tests of FeSe-NC, FeNC, and Pt/C-20%. (e) Performance radar chart of Eonset, E1/2, Tafel slope, jk and jL of FeSe-NC, FeNC and Pt/C-20%. (f) Accelerated degradation tests for the ORR of FeSe-NC. | ||
The electrochemical active surface area (ECSA) was further calculated using the double-layer capacitance (Cdl) derived from CV measurements, aiming to evaluate the available active surface area of the synthesized catalyst (Fig. S22, SI). FeSe-NC exhibits higher Cdl (39.79 mF cm−2) and ECSA (195.21 m2 g−1) than FeNC (12.64 mF cm−2 and 62.01 m2 g−1), favoring the accessibility of active sites along with the improvement of catalytic performance (Table S7, SI). Besides, to explore the actual active sites of FeSe-NC, anionic SCN− poisoning measurements were performed (Fig. S23, SI). As a result, the ORR performance of FeSe-NC is significantly attenuated, which is reflected by an apparent decrease of 38 mV in its E1/2, suggesting obviously that the ORR performance of FeSe-NC is closely related to atomic Fe sites. Stability is another essential factor in evaluating catalytic performance. FeSe-NC displays a current retention of 90% after a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s chronoamperometry test (Fig. S24, SI) and shows no attenuation for E1/2 after accelerated degradation tests (ADTs) (Fig. 4f). The TEM image of FeSe-NC after the ADT indicates that there are no obvious changes in morphology (Fig. S25, SI). The same scenarios were also witnessed in XRD patterns and Raman spectra (Fig. S26, SI). These results identify the superior structural robustness and excellent stability of FeSe-NC.
000 s chronoamperometry test (Fig. S24, SI) and shows no attenuation for E1/2 after accelerated degradation tests (ADTs) (Fig. 4f). The TEM image of FeSe-NC after the ADT indicates that there are no obvious changes in morphology (Fig. S25, SI). The same scenarios were also witnessed in XRD patterns and Raman spectra (Fig. S26, SI). These results identify the superior structural robustness and excellent stability of FeSe-NC.
Site density determination and spin state analysis
In situ scanning electrochemical microscopy (SECM) techniques with micro/nano-scale resolution were employed to explore the local catalytic processes of electrocatalysts in real time and probe their intrinsic catalytic parameters.43,44 The in situ electrochemical cell comprises a sharp platinum ultramicroelectrode (UME) serving as the probe and a concave substrate electrode fabricated by embedding the catalyst into a gold microdisk (Fig. 5a). The tip generation/substrate collection (TG/SC) mode of SECM is a powerful tool to visualize the ORR active region of the catalyst. The essential mechanism of the model is that the probe performs the oxygen evolution reaction at a given potential to produce oxygen. At the same time, the generated oxygen is reduced at various given substrate potentials (Es) by the ORR catalyst in the substrate electrode. The intrinsic catalytic parameters of the catalyst are reflected by collecting information on the change in the response current of the probe to the substrate (Fig. S27–30, SI). When Es is at a high potential, the response current of the substrate is at a low level, indicating that the kinetics of the ORR is low at that moment. Then, as the value of Es gradually decreases, the feedback current gradually increases. Owing to the contribution of the catalyst to the response current, the scanned images of FeSe-NC exhibit higher current values in the center region than in the surrounding regions. When Es = 0.40 V, the feedback current collected from FeSe-NC is 70.2 nA, which is 29.0 nA higher than that from FeNC (Fig. 5b and c). FeSe-NC exhibits a faster increase in feedback current at each Es, as shown in the maximum feedback current curves, highlighting its enhanced intrinsic catalytic activity and kinetics (Fig. S31, SI). | ||
| Fig. 5 (a) Schematic diagram of the TG/SC mode SECM setup. SECM images obtained in TG/SC mode at a substrate potential of 0.4 V (vs. RHE) for (b) FeSe-NC and (c) FeNC. (d) Integrated charge and calculated SD with SI-SECM. (e) Reactivity map based on SD and TOF for FeSe-NC and FeNC (at 0.85 V vs. RHE) and SACs from previous studies. (f) Ultraviolet photoemission spectroscopy spectra of FeSe-NC and FeNC. (g) χm plots for ZFC and computed unpaired 3d electron numbers for FeSe-NC and FeNC. (h) Schematic illustration of the spin-state transfer in FeNC and FeSe-NC. | ||
Accurate measurements of the active site density of catalysts are important to assess the intrinsic reactivity of the active center. Surface interrogation scanning electrochemical microscopy (SI-SECM) has become a strong tool for investigating the behavior of active sites. The technique works on the principle of electrochemical titration in the ferrocene methanol–KOH (FcMeOH-KOH) solution system. Prior to testing, the substrate electrode is subjected to a constant potential to pre-activate the catalyst, during which the Fe sites in the catalyst are oxidized to high valence states (Fe3+). Subsequently, the probe is maintained at an open-circuit potential while a range of given potentials were imposed on the substrate, accompanied by Fe3+ to Fe2+ transition. The substrate is switched to an open circuit at the end of the above process. Equally, as an electrochemical titrant, FcMeOH is oxidized to FcMeOH+ at the probe electrode under constant potential excitation and diffused to the substrate to be reduced by Fe2+. This process records positive feedback current on the probe generated by the FcMeOH+ and FcMeOH transitions. Therefore, the number of active sites actually involved in the ORR was quantified by the quantity of charge (Qtip) collected at the tip (Fig. 5d and S32, SI). Since it is difficult to directly measure the loading of catalyst powder inside the concave electrode by routine methods, we adopted Cdl for indirect determination (Fig. S33, SI). The experimentally determined SDmass value of FeSe-NC is 6.40 × 1020 sites g−1, significantly exceeding that of FeNC (2.45 × 1020 sites g−1). Based on SDmass, the turnover frequency (TOF) of the catalyst was further determined. The results show that the TOF value of FeSe-NC is 0.77 e− site−1 s−1 at 0.85 V vs. RHE, which is superior to that of FeNC (0.56 e− site−1 s−1). The TOF-SD reactivity maps further reveal that FeSe-NC has a jm close to 80.00 A g−1, whereas the jm of FeNC reaches only 24.54 A g−1. Moreover, compared to previously published advanced SACs, FeSe-NC is found to approach the top of the SD-TOF reactivity map (Fig. 5e and Table S8, SI). These findings confirm the favorable intrinsic activity of the Fe–Se dual-atom sites and highlight the validity of the metal–semimetal coupling strategy for enhancing the overall reaction dynamics. In addition, the electronic structure and work function of FeSe-NC and FeNC were explored by utilizing ultraviolet photoelectron spectroscopy (Fig. 5f and S34, SI). The secondary electron cutoff of FeSe-NC is slightly shifted towards higher binding energies. The lower work function of FeSe-NC (6.10 eV) compared to FeNC (6.20 eV) suggests that it requires a lower energy barrier to activate outwardly directed electron transfer to adsorbed oxygen species, which is more favorable for the following ORR steps.
To further unravel the electron configurations of the catalysts, electron paramagnetic resonance (EPR) was performed. FeSe-NC shows stronger EPR signals than FeNC (Fig. S35, SI), confirming that the former contains more unpaired electrons.45 To explain how the neighboring Se affects the FeN4 sites, zero-field cooling temperature-dependent magnetic susceptibility measurements were performed. The electronic structure of FeN4 is stimulated by neighboring Se atoms, thereby enhancing the paramagnetic state of Fe, which can be attributed to the introduction of Se inducing spin splitting of Fe sites.46 By fitting the temperature magnetic susceptibility (M–T) curves (Fig. 5g), the effective magnetic moments (µeff) of FeNC and FeSe-NC are determined to be 0.92µB and 3.60µB, respectively. According to the equation:  , the unpaired electron (n) of the Fe center is further calculated to be 2 for FeSe-NC, higher than that of FeNC, suggesting that Fe sites transform from the low spin (LS) to the middle spin (MS) state after coupling with the Se atom (Fig. 5h and S36, SI). Obviously, the coupling of iron to selenium generates more unpaired electrons, thus stimulating the Fe sites to develop higher spin states. The transformation facilitates the optimization of O2 activation/dissociation and thus boosts catalytic activity.47
, the unpaired electron (n) of the Fe center is further calculated to be 2 for FeSe-NC, higher than that of FeNC, suggesting that Fe sites transform from the low spin (LS) to the middle spin (MS) state after coupling with the Se atom (Fig. 5h and S36, SI). Obviously, the coupling of iron to selenium generates more unpaired electrons, thus stimulating the Fe sites to develop higher spin states. The transformation facilitates the optimization of O2 activation/dissociation and thus boosts catalytic activity.47
DFT analysis
To reveal the detailed ORR mechanism on FeSe-NC, DFT calculations were further performed. According to the XANES and EXAFS fitting results, three models were constructed and compared: FeN4, FeN3Se1, and FeN3Se2 (Fig. 6a). The four-electron reduction mechanism under alkaline conditions was employed for Gibbs free energy calculations (Fig. 6b and S37, SI). At U = 0.46 V, the rate-determining step (RDS) for the FeN4 model is found to be the *OOH adsorption step with an overpotential of 0.95 V. The RDS for both the FeN3Se1 and FeN3Se2 systems is the *OH desorption step with overpotentials of 1.00 V and 0.89 V, respectively. Furthermore, from a perspective of geometrical configuration, FeN3Se2 lacks the larger tensile tetragonal structure, making it a more representative structure of experimentally synthesized materials. The lower overpotential indicates that FeN3Se2 has faster ORR kinetics than FeN4, which shows good agreement with ORR experimental results. Hence, we can reasonably assume that the coupling of Fe–Se dual-atom sites can decrease the reaction overpotential and enhance ORR activity. | ||
| Fig. 6 (a) Theoretical analysis on FeN4, FeN3Se1, and FeN3Se2 models. (b) Free energy diagrams of FeN4, FeN3Se1, and FeN3Se2 at U = 0 V and U = 0.46 V. (c) The contour mapping of in situ ATR-SEIRAS spectra of FeSe-NC under various potentials. (d) Charge density differences of FeN3Se2 and FeN4. Electron density accumulation is represented by the blue iso-surface, and electron density loss is depicted by the yellow iso-surface. (e) DOS of FeN4 and FeN3Se2. | ||
Dynamic structural evolution exerts a significant influence on the catalytic behavior of the catalysts. By means of in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), the dynamic evolution of the FeSe-NC active sites was thoroughly disclosed. The peak located at 3450 cm−1 is attributed to the stretching vibrational mode of the adsorbed OH group (Fig. 6c). The enhanced peak intensity under increasing overpotential implies that OH desorption from the catalytic site was impeded,48 which is in agreement with DFT simulations.
The electronic effects and synergistic interactions between Fe and Se were further examined using differential charge density distributions and the density of states (DOS). Electronic interactions within the Fe–Se dual-atom pairs lead to charge redistribution, where the electron-donating tendency of Se induces a directional transfer of electrons to adjacent FeN4, thus disrupting the original symmetric electron-density configuration of FeN4. In parallel, the Bader charge of Fe in the FeN3Se2 model is larger than that in the FeN4 model, which facilitates the activation of ORR intermediates (Fig. 6d). As the d-band center is thought to be closely related to metal-intermediate interactions, more insights can be obtained by studying the electronic structure of the Fe center.49 The d-band center of the Fe DOS in the FeN3Se2 model is upshifted compared with the FeN4 model, suggesting that Fe sites in the FeN3Se2 model have more activated electrons to react with intermediates (Fig. 6e and S38, SI). DFT calculations suggest that the coupling of Fe–Se dual-atoms can break the symmetric electron distribution in FeN4 to upshift the d-band center, which can facilitate the activation of oxygen intermediates and thus accelerate the ORR kinetics.
The development of high-energy-density ZABs capable of scalable production critically relies on high-performance air cathodes. To address this requirement, we demonstrate a roll-to-roll fabrication strategy for an integrated electrode, as schematically illustrated in Fig. 7a. Briefly, a homogeneous slurry consisting of the catalyst, poly(tetrafluoroethylene) (PTFE) binder and acetylene black was first rolled into a freestanding film, which was then laminated onto a nickel mesh current collector. This lamination procedure was repeated to build a robust air cathode with strong interfacial adhesion. After drying, a final rolling step was applied to enhance mechanical stability and ensure structural integrity. Compared to conventional wet-spray coating, this process offers distinct advantages. Firstly, the use of low-cost PTFE, rather than expensive Nafion, as a binder significantly reduces electrode manufacturing costs.
 | ||
| Fig. 7 (a) Schematic synthesis of integrated cathodes by the roll-to-roll method. (b) A digital photo of a large-scale electrode (150 cm × 11 cm) manufactured via the roll-to-roll synthesis method. (c) Discharging polarization curves and power plots of bipolar stacking ZABs. (d). Comparison of power and open-circuit voltage for bipolar stacking ZABs assembled with air cathodes of different catalyst loadings. (e) Rate performance of the bipolar stacking ZABs. (f) Galvanostatic discharge performance of ampere-hour-scale ZABs at 1.0 A. (g) Galvanostatic charge–discharge cycling curves of bipolar stacking and conventional ZABs at 1.0 A. | ||
Secondly, the intensive homogenization of the precursor slurry ensures that the catalyst, conductive agent, and binder are intimately mixed, facilitating the formation of a well-defined and coherent catalytic layer. This structural homogeneity, further enhanced by the repeated rolling operations, promotes dense packing and intimate interfacial contact within the electrode, thereby effectively reducing interfacial impedance. Electrochemical impedance measurements performed at three distinct locations on the same electrode reveal remarkably low and comparable resistance values (Fig. S39 and 40, SI). This spatial uniformity in electrical response provides cogent proof of a highly homogeneous and dense catalytic layer. The SEM image demonstrates a consistent and uniform distribution of the FeSe-NC catalysts, acetylene black, and PTFE binder, which is corroborated by elemental mapping (Fig. S41, SI). The XRD pattern (Fig. S42, SI) shows the characteristic peaks of the three components, with no extra phases detected. This confirms their successful physical integration and indicates no detectable chemical reaction occurred between them. Finally, employing nickel mesh as the current collector enhances the electrode's capability to withstand high current, a critical factor for practical ampere-hour-level device applications. The combination of these merits with the inherent scalability of the roll-to-roll process has enabled the successful fabrication of large-area air cathodes (Fig. 7b).
Performance of ZABs
To systematically evaluate the impact of battery configuration on the performance, we assembled bipolar stacking and conventional ZABs for a comparative study using the as-fabricated air cathodes. The bipolar stacking cell was constructed with free-standing zinc foil as the central anode, flanked by liquid electrolytes and our self-made air cathodes on both sides (Fig. S43 and S44, SI). This battery configuration can be viewed as an equivalent parallel circuit, where the doubled active area directly boosts current and power output. This structural advantage works synergistically with promoted reaction kinetics and uniform zinc deposition to concurrently improve zinc utilization, suppress side reactions, and reduce overpotential. Therefore, a simultaneous improvement in rate capability, energy capacity, and cycling stability can be achieved. To investigate the optimal catalyst loading, conventional ZABs were assembled using air cathodes with different catalyst loadings. The ZABs driven by the air electrode with 5 mg cm−2 catalyst loading exhibit a high open-circuit voltage (OCV) of 1.54 V and a maximum power of 2.3 W, outperforming those with loadings of 3 and 7 mg cm−2 (Fig. S45–47, SI). The obtained electrode was then extended to a bipolar stacking cell configuration for a direct comparison. The bipolar stacking cell architecture universally enhanced the performance of air cathodes relative to the conventional design. It not only delivered consistently higher power outputs across all catalyst loadings tested, but also achieved a maximum power output of 3.5 W at an optimal loading of 5 mg cm−2, demonstrating its comprehensive performance advantage over conventional designs (Fig. 7c, d and S48, SI). To assess the rate capability, the discharge voltage profiles were systematically compared (Fig. 7e). Obviously, bipolar stacking ZABs maintain a superior discharge voltage platform compared to conventional ZABs across a range of current. Moreover, benefiting from the good stability of FeSe-NC and air cathodes, it can be found that the discharge potential of ZABs can return to the initial stage after a high discharge rate of 1.5 A, confirming its superior rate performance and reversibility. To validate the practical potential of the electrodes, the ZABs with both configurations were subjected to galvanostatic discharge at 1.0 A. Following different discharge periods, the bipolar stacking and conventional cells delivered capacities of 6.09 Ah and 3.94 Ah per cell, respectively (Fig. 6f). These results successfully demonstrate the feasibility of constructing ZABs with ampere-hour-level discharge capacity, while clearly indicating that the bipolar stacking structure holds significant advantages for long-duration energy storage applications. The galvanostatic cycling of ampere-hour-scale ZABs was studied under galvanostatic discharge/charge conditions at 1.0 A (Fig. 7g). The bipolar stacking ZAB showed impressive performance, retaining stable voltage efficiency for over 60 h and significantly exceeding the 28 h duration of the conventional cell. These properties highlight the competitive performance of our Ah-level ZAB among recent studies (Table S9, SI). These results demonstrate that air electrodes fabricated via the roll-to-roll process exhibit exceptional structural stability and can meet the requirements for high power and ampere-hour-level capacity output demanded by ZABs, showing great promise for practical energy storage applications. Moreover, the bipolar stacking configuration not only unlocks the practical application potential of self-made air electrodes in energy storage but also provides a valuable strategy for constructing highly efficient ZAB systems.Conclusions
In summary, this work demonstrates a holistic design strategy for advanced Zn–air batteries through the integrated optimization of catalyst engineering, electrode fabrication, and battery architecture. First, the development of a synchronous complexation–polymerization approach enables the precise construction of Fe–Se dual-atom catalysts with a unique metal–semimetal coordination configuration, where Se introduction induces asymmetric charge distribution on Fe sites and induces a medium-spin state transition, collectively optimizing the bonding behavior with oxygen intermediates for enhanced reaction kinetics. Second, the establishment of a scalable roll-to-roll process facilitates the fabrication of integrated air cathodes, featuring highly uniform catalytic layers and significantly reduced interfacial resistance. Finally, the implementation of a bipolar stacking cell configuration substantially improves battery performance, with the assembled ampere-hour-scale devices delivering a maximum power output of 3.5 W, a large capacity of 6.09 Ah, and extended cycling stability exceeding 60 h at 1.0 A. This work demonstrates the effectiveness of a multi-level design strategy that seamlessly connects atomic-scale catalyst engineering, macroscopic electrode processing, and device-level configuration optimization, offering a viable pathway toward practical Zn–air battery systems.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Y. Wei, K. Yuan, and Y. Chen designed the project. Y. Wei, J. Huang, and L. Li performed the syntheses, physical characterization studies, and electrochemical tests. Y. Wei, X. Zhuang, C. Lu, and D. Lützenkirchen–Hecht tested and analyzed the XANES and EXAFS. Y. Ma and X. J. Gao performed the DFT calculations. All authors participated in the discussion of experimental and calculation results.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of experimental methods and characterisation data. See DOI: https://doi.org/10.1039/d5sc08392c.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (52573228, 52373187, and 22467013), the National Youth Top-notch Talent Support Program of China, the Natural Science Foundation of Jiangxi Province (20224ACB204006), the “Double Thousand Plan” Science and Technology Innovation High-End Talent Project of Jiangxi Province (jxsq2023201094), and the Gan Poyang talents support program, academic and technical leaders of major disciplines (20232BCJ23014).Notes and references
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- R. F. Service, Science, 2021, 372, 890–891 CrossRef CAS PubMed.
- P. Jiang, Y. Xu, Z. Gong, B. Ge, L. Ding, C. Huang, X. Qiu and Z. Pei, Angew. Chem., Int. Ed., 2025, 64, e202504188 CrossRef CAS.
- Z. Zhuang, Y. Li, R. Yu, L. Xia, J. Yang, Z. Lang, J. Zhu, J. Huang, J. Wang, Y. Wang, L. Fan, J. Wu, Y. Zhao, D. Wang and Y. Li, Nat. Catal., 2022, 5, 300–310 CrossRef CAS.
- W. Zhou, B. Li, X. Liu, J. Jiang, S. Bo, C. Yang, Q. An, Y. Zhang, M. A. Soldatov, H. Wang, S. Wei and Q. Liu, Nat. Commun., 2024, 15, 6650 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS.
- L. Li, X. Tang, S. Huang, C. Lu, D. Lützenkirchen-Hecht, K. Yuan, X. Zhuang and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202301642 CrossRef CAS PubMed.
- A. Mehmood, M. Gong, F. Jaouen, A. Roy, A. Zitolo, A. Khan, M.-T. Sougrati, M. Primbs, A. M. Bonastre, D. Fongalland, G. Drazic, P. Strasser and A. Kucernak, Nat. Catal., 2022, 5, 311–323 CrossRef CAS.
- D. Menga, J. L. Low, Y.-S. Li, I. Arčon, B. Koyutürk, F. Wagner, F. Ruiz-Zepeda, M. Gaberšček, B. Paulus and T.-P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- C. Han, S. Zhang, H. Zhang, Y. Dong, P. Yao, Y. Du, P. Song, X. Gong and W. Xu, eScience, 2024, 4, 100269 CrossRef.
- Y. Li, H. Sun, L. Ren, K. Sun, L. Gao, X. Jin, Q. Xu, W. Liu and X. Sun, Angew. Chem., Int. Ed., 2024, 63, e202405334 CrossRef CAS.
- B. Huang, S. Huang, C. Lu, L. Li, J. Chen, T. Hu, D. Lützenkirchen-Hecht, K. Yuan, X. Zhuang and Y. Chen, CCS Chem., 2022, 5, 1876–1887 CrossRef.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2021, 12, 2102715 CrossRef.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- J. Du, G. Han, W. Zhang, L. Li, Y. Yan, Y. Shi, X. Zhang, L. Geng, Z. Wang, Y. Xiong, G. Yin and C. Du, Nat. Commun., 2023, 14, 4766 CrossRef CAS.
- H. Hu, J. Wang, K. Liao, Z. Chen, S. Zhang, B. Sun, X. Wang, X. Ren, J. Lin and X. Han, Adv. Mater., 2024, 37, 2416126 CrossRef.
- Y. Liu, J. Li, Z. Lv, H. Fan, F. Dong, C. Wang, X. Chen, R. Liu, C. Tian, X. Feng, W. Yang and B. Wang, J. Am. Chem. Soc., 2024, 146, 12636–12644 CrossRef CAS.
- L. Lei, X. Guo, X. Han, L. Fei, X. Guo and D.-G. Wang, Adv. Mater., 2024, 36, 2311434 CrossRef CAS.
- Y. Wang, P. Meng, Z. Yang, M. Jiang, J. Yang, H. Li, J. Zhang, B. Sun and C. Fu, Angew. Chem., Int. Ed., 2023, 62, e202304229 CrossRef CAS.
- B. Tang, Y. Zhou, Q. Ji, Z. Zhuang, L. Zhang, C. Wang, H. Hu, H. Wang, B. Mei, F. Song, S. Yang, B. M. Weckhuysen, H. Tan, D. Wang and W. Yan, Nat. Synth., 2024, 3, 878–890 CrossRef CAS.
- Q. Jing, Z. Mei, X. Sheng, X. Zou, Q. Xu, L. Wang and H. Guo, Adv. Funct. Mater., 2024, 34, 2307002 CrossRef CAS.
- Y. Li, H. Wang, C. Chen, X. Xie, Y. Yang, X. Tan, K. Jiang, N. Chen, H. Zhang and Z. Li, Nat. Commun., 2025, 16, 8085 CrossRef CAS.
- J. Mun, T. Song, M.-S. Park and J. H. Kim, Adv. Mater., 2025, 37, 2506123 CrossRef CAS.
- Y. Lin, M. Wu, J. Sun, L. Zhang, Q. Jian and T. Zhao, Adv. Energy Mater., 2021, 11, 2101612 CrossRef CAS.
- A. Lee, J. H. Lee and S. H. Ahn, Adv. Funct. Mater., 2025, 35, 2414164 CrossRef CAS.
- S. S. Shinde, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, H.-D. Um, S. U. Lee and J.-H. Lee, Adv. Mater., 2023, 35, 2303509 CrossRef CAS.
- Z. Pei, L. Ding, C. Wang, Q. Meng, Z. Yuan, Z. Zhou, S. Zhao and Y. Chen, Energy Environ. Sci., 2021, 14, 4926–4935 RSC.
- T. Wang, Q. Wang, Y. Wang, Y. Da, W. Zhou, Y. Shao, D. Li, S. Zhan, J. Yuan and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13466–13471 CrossRef CAS.
- X. Meng, C. Yu, X. Song, J. Iocozzia, J. Hong, M. Rager, H. Jin, S. Wang, L. Huang, J. Qiu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 4682–4686 CrossRef CAS.
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 135, e202219191 CrossRef.
- Y. Wei, M. Huang, Y. Wu, X. Tang, K. Yuan and Y. Chen, Adv. Funct. Mater., 2024, 34, 2404787 CrossRef CAS.
- M. Wu, J. Wang, Z. Liu, X. Liu, J. Duan, T. Yang, J. Lan, Y. Tan, C. Wang, M. Chen and K. Ji, Adv. Mater., 2023, 35, 2209924 CrossRef CAS.
- J. Kim, K. Park, M. Kim, H. Lee, J. Choi, H. B. Park, H. Kim, J. Jang, Y.-H. Kim, T. Song and U. Paik, Adv. Energy Mater., 2024, 14, 2303455 CrossRef CAS.
- P. Du, B. Deng, X. He, W. Zhao, H. Liu, Y. Long, Z. Zhang, Z. Li, K. Huang, K. Bi, M. Lei and H. Wu, ACS Nano, 2025, 19, 1327–1339 CrossRef CAS.
- L. Li, J. Zhu, F. Kong, Y. Wang, C. Kang, M. Xu, C. Du and G. Yin, Matter, 2024, 7, 1517–1532 CrossRef CAS.
- B. Huang, X. Tang, Y. Hong, L. Li, T. Hu, K. Yuan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202306667 CrossRef CAS.
- Y. Ma, X. Meng, K. Li, L. Zhang, Y. Du, X. Cai and J. Qiu, ACS Catal., 2023, 13, 1290–1298 CrossRef CAS.
- I. Martinaiou, A. H. A. Monteverde Videla, N. Weidler, M. Kübler, W. D. Z. Wallace, S. Paul, S. Wagner, A. Shahraei, R. W. Stark, S. Specchia and U. I. Kramm, Appl. Catal., B, 2020, 262, 118217 CrossRef CAS.
- H. Hu, J. Wang, B. Cui, X. Zheng, J. Lin, Y. Deng and X. Han, Angew. Chem., Int. Ed., 2022, 61, e202114441 CrossRef CAS.
- J. Chen, B. Huang, R. Cao, L. Li, X. Tang, B. Wu, Y. Wu, T. Hu, K. Yuan and Y. Chen, Adv. Funct. Mater., 2022, 23, 2209315 Search PubMed.
- Z. Sun, H. Zhang, L. Cao, X. Liu, D. Wu, X. Shen, X. Zhang, Z. Chen, S. Ru, X. Zhu, Z. Xia, Q. Luo, F. Xu and T. Yao, Angew. Chem., Int. Ed., 2023, 62, e202217719 CrossRef CAS.
- C. Santana Santos, B. N. Jaato, I. Sanjuán, W. Schuhmann and C. Andronescu, Chem. Rev., 2023, 123, 4972–5019 CrossRef CAS.
- E. C. R. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2021, 122, 3292–3335 CrossRef.
- W. Ren, X. Tan, C. Jia, A. Krammer, Q. Sun, J. Qu, S. C. Smith, A. Schueler, X. Hu and C. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202203335 CrossRef CAS.
- L. Wang, Z. Mei, Q. An, X. Sheng, Q. Jing, W. Huang, X. Wang, X. Zou and H. Guo, Chem Catal., 2023, 3, 100758 CAS.
- X. Wei, S. Song, W. Cai, X. Luo, L. Jiao, Q. Fang, X. Wang, N. Wu, Z. Luo, H. Wang, Z. Zhu, J. Li, L. Zheng, W. Gu, W. Song, S. Guo and C. Zhu, Chem, 2023, 9, 181–197 CAS.
- P. Zhang, H.-C. Chen, H. Zhu, K. Chen, T. Li, Y. Zhao, J. Li, R. Hu, S. Huang, W. Zhu, Y. Liu and Y. Pan, Nat. Commun., 2024, 15, 2062 CrossRef CAS.
- X. Wang, Y. An, L. Liu, L. Fang, Y. Liu, J. Zhang, H. Qi, T. Heine, T. Li, A. Kuc, M. Yu and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209746 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |