
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to allyl-gem-difluorides via allylation and Cope rearrangement
Haeun
Kim†
 ,
Seungcheol
Han†
,
Yunhui
Jang
,
Yujin
Jung
and
Seung Hwan
Cho
*
,
Seungcheol
Han†
,
Yunhui
Jang
,
Yujin
Jung
and
Seung Hwan
Cho
*
Department of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang, 37673, Rep. of Korea. E-mail: seunghwan@postech.ac.kr
First published on 3rd December 2025
Abstract
Fluorinated molecules play crucial roles in medicinal and materials chemistry, as the presence of fluorine profoundly alters molecular electronics and reactivity. Among them, allyl gem-difluorides have attracted growing attention as versatile synthetic intermediates, but their selective construction remains challenging. Here, we disclose an efficient strategy for synthesizing allyl gem-difluorides bearing (E)-vinyl boronic esters through a LiOtBu-mediated deborylative allylation/Cope rearrangement sequence of 3-boryl-1,1-difluoro allylboronic ester with allyl bromides. Four competing pathways (α-SN2, α-SN2′, γ-SN2, and γ-SN2′) were identified: α-addition generates intermediates that undergo Cope rearrangement, while γ-addition directly delivers the same products. Mechanistic studies, supported by control experiments as well as NBO and FMO analyses, demonstrate a synergistic effect between the 1,1-difluoro group and the boronic ester that lowers the activation barrier of the rearrangement. This transformation displays broad substrate scope and provides versatile building blocks for the construction of allyl gem-difluorides.
Introduction
Fluorine-containing organic molecules play pivotal roles in agrochemicals, pharmaceuticals, and advanced materials owing to their distinctive physicochemical properties.1 Among fluorinated motifs, the difluoromethylene (CF2) unit serves as a valuable bioisostere of oxygen and carbonyl groups, imparting unique features such as altered dipole moments, increased acidity of adjacent bonds, and characteristic conformational preferences (Scheme 1a).2 These attributes often translate into enhanced metabolic stability and biocompatibility in drug-like molecules.3 Within this context, allyl gem-difluorides represent a particularly intriguing class of molecules because of their unique chemical reactivity and potential applications in the design of bioactive compounds.4,5 | ||
| Scheme 1 Approaches for synthesizing allyl gem-difluorides. Bpin: (pinacolato)boryl. | ||
Over the past few decades, various approaches have been developed for the synthesis of allyl gem-difluorides.6,7 Among these, 1,1-difluoro allylmetalloids have proven to be versatile building blocks for the incorporation of a difluoromethylene unit (Scheme 1b). These reagents have enabled Pd-catalyzed gem-difluoroallylation of aryl halides8 as well as transition-metal free allylation of carbonyls and imines,9 thereby providing efficient access to allyl gem-difluorides. Nevertheless, the use of 1,1-difluoro allylmetalloids has so far been applied primarily to the synthesis of allyl gem-difluorides bearing terminal alkenes. Transformations affording allyl gem-difluorides bearing a 1,2-disubstituted internal alkene unit are rare9f,m and often proceed with poor E/Z selectivity.8b,9b Therefore, the development of a new 1,1-difluoro allylmetalloid reagent and a complementary synthetic platform capable of delivering allyl gem-difluorides bearing 1,2-disubstituted internal alkenes with high stereocontrol remains a significant challenge.
As part of our ongoing research on gem-diboron chemistry,10 we envisioned that incorporating fluorinated motifs into gem-diboryl frameworks11 would provide an effective platform for accessing fluorine-containing molecules. Herein, we report a strategy for synthesizing allyl gem-difluorides bearing (E)-vinyl boronic esters (Scheme 1c). A newly synthesized 3-boryl-1,1-difluoro allylboronic ester undergoes LiOtBu-mediated deborylative allylation with allyl bromides through two distinct pathways: α-allylation, which produces intermediates that undergo Cope rearrangement,12 and γ-allylation, which directly affords the same products. Mechanistic investigations demonstrate that the combined effects of the 1,1-difluoro substituents and the boronic ester significantly lower the activation barrier of the rearrangement, enabling efficient and selective access to diverse allyl gem-difluorides.
Results and discussion
We first hypothesized that if we could synthesize 3-boryl-1,1-difluoro allylboronic ester, it could serve as a versatile synthon for the synthesis of structurally diverse allyl gem-difluorides via deborylative C–C bond formation with allyl electrophiles. To realize this concept, we first developed a method for preparing the target 3-boryl-1,1-difluoro allylboronic ester. Drawing inspiration from our previous work on palladium-catalyzed cross-coupling of organo(pseudo)halides with diborylmethylzinc halides,13 we applied related conditions to this system. When 2,2-difluorovinyl p-toluenesulfonate (1) and diborylmethylzinc halide (2) were subjected to Pd(TFA)2 catalysis with RuPhos as the ligand in THF at 80 °C, the desired 3-boryl-1,1-difluoro allylboronic ester (3) was obtained in 90% yield on a gram scale (1.5 g, Scheme 2a).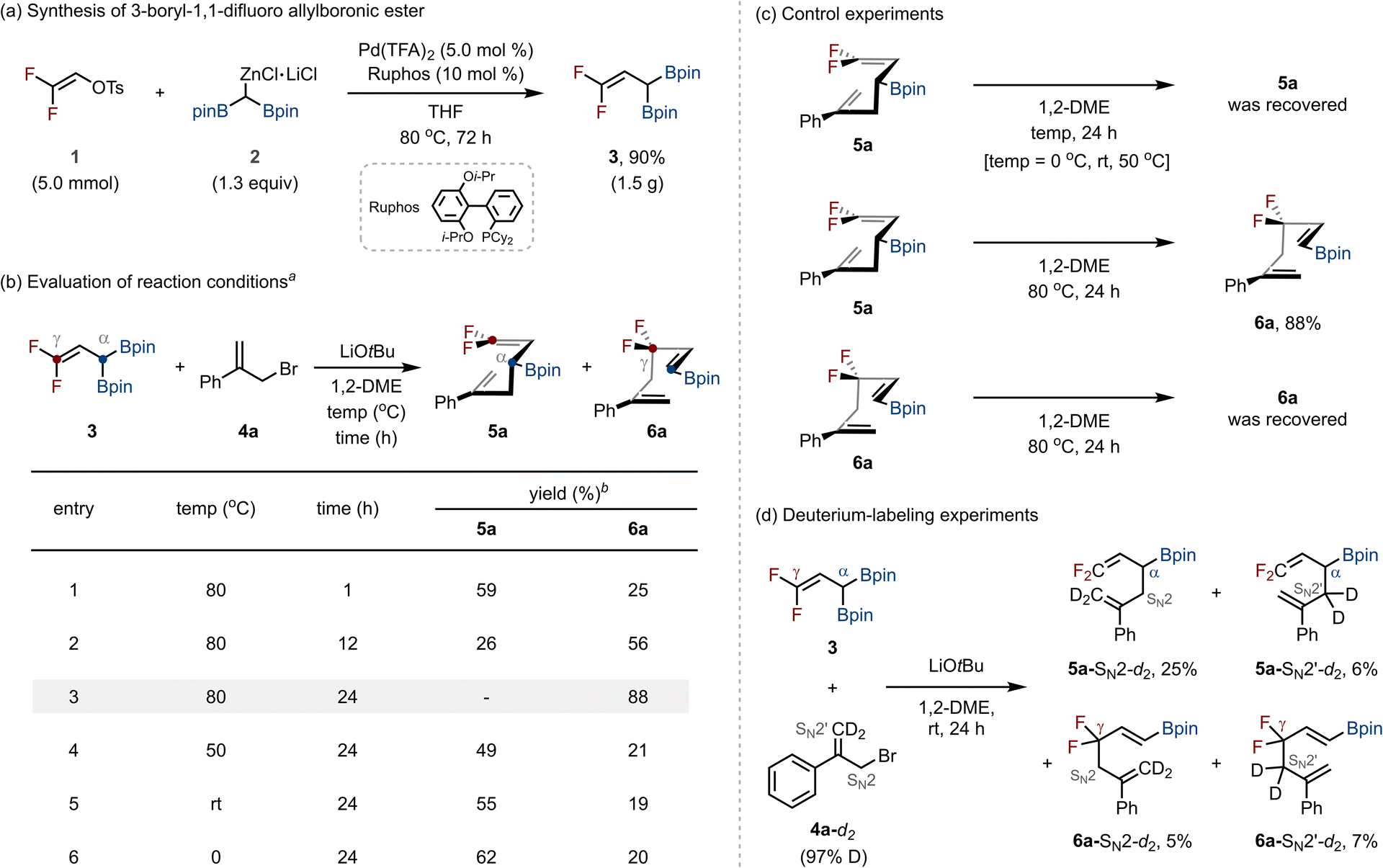 | ||
| Scheme 2 Reaction discovery. a3 (0.22 mmol), 4a (0.10 mmol), and LiOtBu (0.22 mmol) in 1,2-DME (0.20 M) at the indicated temperature and time. The 1H NMR yield was determined using CH2Br2 as an internal standard. TFA: trifluoroacetate; i-Pr: isopropyl; Cy: cyclohexyl; THF: tetrahydrofuran; 1,2-DME: 1,2-dimethoxyethane; Ph: phenyl. | ||
With compound 3 in hand, we next investigated its reactivity in base-promoted deborylative C–C bond-forming reactions14 with allyl electrophiles (Scheme 2b). As a model study, (3-bromoprop-1-en-2-yl)benzene (4a) was reacted with 3 under LiOtBu in 1,2-dimethoxyethane (1,2-DME) at 80 °C. After 1 h, a mixture of the α-addition product 5a (59%) and the γ-addition product 6a (25%) was obtained, with 5a as the major product (entry 1). Extending the reaction time to 12 h decreased the yield of 5a (26%) while increasing that of 6a (56%, entry 2). After 24 h, 6a was formed exclusively in 88% yield (entry 3). Lowering the reaction temperature led to the formation of a mixture of 5a and 6a, with 5a as the major product (entries 4–6).
To confirm whether 6a arises from the Cope rearrangement of 5a, we conducted control experiments (Scheme 2c). When pure 5a was subjected to the reaction conditions at 0–50 °C, no conversion occurred. At 80 °C, however, 5a was quantitatively transformed into 6a. In contrast, subjecting 6a to the same conditions resulted in no reversion to 5a. The optimization studies and control experiments indicated that the LiOtBu-mediated coupling of 3 with 4a proceeds via two competing pathways: γ-addition, which directly furnishes 6a, and α-addition, which yields 5a that undergoes irreversible Cope rearrangement to 6a at elevated temperature. To further probe the pathway, deuterium-labeling experiments were performed with 3 and deuterium-labeled allyl bromide 4a-d2 at room temperature (Scheme 2d). In this reaction, four distinct products were obtained, 5a-SN2-d2 (25%), 5a-SN2′-d2 (6%), 6a-SN2-d2 (5%), and 6a-SN2′-d2 (7%), showing that the reaction can proceed through all four possible pathways: nucleophilic attack from either the α- or γ-position of 3, occurring via SN2 or SN2′ displacement on allyl bromide 4a.
To gain deeper insight into the reaction pathways, we performed density functional theory (DFT) calculations at the SMD (THF) ωB97X-D4/def2-TZVPP//B3LYP/6-31G* level of theory (Scheme 3). THF was chosen as the model solvent, rather than 1,2-DME, to account for possible solvent coordination effects while reducing computational cost and complexity. In the presence of LiOtBu, 3-boryl-1,1-difluoro allylboronic ester 3 generates a boronate species that undergoes C–B bond cleavage to form the s-trans carbanion INT-1, which can isomerize to s-cis carbanion INT-2 (see the SI for details). Both intermediates are competent for allylation with allyl bromide 4a, either at the α- or γ-position, and each can proceed via SN2 or SN2′ displacement, which gives rise to four possible pathways: α-SN2, α-SN2′, γ-SN2, and γ-SN2′. The calculations revealed distinct conformational preferences. Allylation at the α-position of the carbanion (α-SN2 and α-SN2′) proceeds preferentially through INT-2, with lower activation barriers (TS-2-A: 16.63 kcal mol−1; TS-2-B: 17.11 kcal mol−1) than through INT-1 (TS-1-A: 20.87 kcal mol−1; TS-1-B: 19.18 kcal mol−1). In contrast, γ-allylation (γ-SN2, γ-SN2′) is favored viaINT-1 (TS-1-C: 17.29 kcal mol−1; TS-1-D: 17.81 kcal mol−1) over INT-2 (TS-2-C: 18.87 kcal mol−1; TS-2-D: 19.48 kcal mol−1). Overall, TS-2-A (α-SN2 from INT-2) was identified as the most favorable transition state, followed by TS-2-B, TS-1-C, and TS-1-D. Taken together, these results indicate that all four pathways are energetically accessible, in full agreement with the deuterium-labeling experiments that revealed the participation of each route, and they provide the foundation for analyzing the subsequent Cope rearrangement step. Because allyl bromide 4a is symmetric, both SN2 and SN2′ pathways at a given position lead to the same product. The γ-addition pathways directly deliver 6a, whereas the α-addition pathways first generate conformer 5a′, which relaxes to 5a by bond rotation and then undergoes Cope rearrangement via a chair-like transition state (TS-3) to yield the thermodynamically favored product 6a. These computational findings align well with the experimental selectivities summarized in Scheme 2b.
 | ||
| Scheme 3 Energy profile for the allylation and Cope rearrangement. | ||
We further examined the origin of stereoselectivity in the Cope rearrangement, as only (E)-vinylboronic esters were observed. Calculations revealed a substantial energy difference between the competing transition states: TS-3 leading to the (E)-isomer had a lower activation energy (−19.01 kcal mol−1), whereas that of TS-3-(Z) was significantly higher (−14.25 kcal mol−1). The 4.76 kcal mol−1 gap arises from steric interactions, as visualized in Newman projections: in TS-3-(Z), severe steric repulsion occurs between the aryl substituent and the Bpin group, whereas TS-3 allows a more favorable spatial arrangement. As a result, the rearrangement proceeds predominantly viaTS-3, accounting for the exclusive (E)-selectivity observed experimentally.
Next, the potential roles of fluorine and boron substituents in the Cope rearrangement of 5a to 6a were examined through a series of control experiments (Scheme 4a). When allyl boronic ester 7, lacking fluorine substituents, was subjected to the standard conditions, the rearranged product 8 was obtained in only 16% yield. In the case of compound 9, a derivative of 5a without the Bpin group, product 10 was formed in merely 9% yield, with 91% of the starting material recovered. To further probe the influence of the boron substituent, we tested compound 11, bearing a CH2Bpin group instead of Bpin, which furnished product 12 in only 30% yield. These results strongly indicate that both the geminal difluoro substituents and the Bpin moiety are essential, together exerting a cooperative effect that promotes the Cope rearrangement.
 | ||
| Scheme 4 Control experiments and computational mechanistic studies. | ||
Further computational studies were undertaken to gain a more detailed understanding of the Cope rearrangement. To probe the role of the Bpin group,15 we compared compounds 5a and 11, focusing on their transition states TS-3 and TS-3′ (Scheme 4b). The activation barrier for TS-3 (33.23 kcal mol−1) was substantially lower than that for TS-3′ (36.13 kcal mol−1), highlighting the influence of the Bpin substituent. Natural bond orbital (NBO) analysis16 provided insight into this difference. In TS-3, the σ (C3–C4) bond is aligned with the vacant p orbital of boron [p(B)], enabling efficient σ → p(B) electron delocalization and leading to a strong NBO interaction (E2 = 6.48 kcal mol−1). By contrast, in TS-3′ the σ (C3–C4) bonding orbital is aligned with the σ* (C7–B) antibonding orbital, exhibiting a weaker interaction (E2 = 1.84 kcal mol−1). This enhanced delocalization in TS-3 reduces the σ (C3–C4) bond occupancy (1.777e vs. 1.823e in TS-3′) and elongates the bond (1.818 Å), thereby weakening the C3–C4 bond and lowering the barrier for rearrangement.
Building upon our investigation of the influence of the boron unit, we next examined the role of fluorine substituents17 by conducting frontier molecular orbital (FMO) analysis18 of 5a and its non-fluorinated analogue 7 (Scheme 4c). For the difluorinated substrate 5a, the π orbital of the internal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond corresponds to the HOMO−1,19 while the π* orbital of the terminal CC bond corresponds to the LUMO. Comparison with 7 (X = H) revealed a reduced [HOMO−1]–[LUMO] gap in 5a, arising from an elevation of the HOMO−1 energy by 0.12 eV. This electronic perturbation is attributed to the mesomeric electron-donating contribution of the fluorine substituents via p-π conjugation20 (see the SI for details).
C bond corresponds to the HOMO−1,19 while the π* orbital of the terminal CC bond corresponds to the LUMO. Comparison with 7 (X = H) revealed a reduced [HOMO−1]–[LUMO] gap in 5a, arising from an elevation of the HOMO−1 energy by 0.12 eV. This electronic perturbation is attributed to the mesomeric electron-donating contribution of the fluorine substituents via p-π conjugation20 (see the SI for details).
With this mechanistic understanding, we next examined the substrate scope of allyl bromides 4 in the developed allylation/Cope rearrangement sequence (Table 1). A range of C2-aryl-substituted allyl bromides bearing electron-neutral, electron-donating, or electron-withdrawing groups at the para-position of the phenyl ring furnished products 6a–6h in excellent yields. Substituents at the meta- and ortho-positions of the C2-phenyl group were also tolerated, delivering products 6i–6k, although 6k was obtained only in low yield even at elevated temperature. Allyl bromides having 3,5-dimethylphenyl, protected catechol, naphthyl, and benzofuran groups also underwent the reaction smoothly to give 6l–6o in low to good yields. Of note, para-methoxy- and naphthyl-substituted allyl bromides (4b and 4n) exhibited thermal sensitivity, which was addressed by performing the initial allylation with 3 at 0 °C for 24 h, followed by heating to 80 °C for 12 h. Beyond aryl substitution, allyl bromides bearing aliphatic groups at the C2 position also proved viable, although higher temperatures were required. Substrates with methyl, hexyl, or cyclohexyl groups furnished 6p–6r in moderate yields. Allyl bromides containing protected amines and tert-butyldimethylsilyl (TBS) ethers also participated, affording 6s and 6t, respectively. In contrast, cinnamyl bromide underwent only the initial allylation to give 1,1-difluorinated 1,5-hexadiene 5u, without subsequent Cope rearrangement, even under forcing conditions (150 °C, 24 h). This outcome is likely due to the energetic penalty associated with disrupting π-conjugation and steric hindrance from the phenyl group.

The obtained allyl gem-difluorides bearing (E)-vinyl boronic esters serve as valuable precursors for synthesizing diverse gem-difluoro-containing molecules. To demonstrate the scalability and practicability of this method, a gram-scale reaction was performed (Scheme 5). Treatment of 4a (5.0 mmol) with 3 (2.2 equiv) furnished 6a in 60% yield after extended reaction time. It should be noted that the crude product could be used directly in subsequent transformations without purification. Protodeboronation of 6a with a stoichiometric amount of AgF in THF/D2O/MeOD at 80 °C afforded product 13 in 55% yield with >99% deuterium incorporation.21 Palladium-catalyzed Suzuki–Miyaura cross-coupling of the vinyl Bpin moiety of 6a with β-bromostyrene gave product 14 in 80% yield,22 while an analogous cross-coupling of 6a with an estrone-derived triflate afforded 15 in 81% yield.23 Hydrogenation of the alkenes in 6a in the presence of 10 mol% Pd/C in MeOH at 60 °C under an H2 atmosphere (1 atm) yielded the corresponding gem-difluoroalkyl molecule containing a terminal Bpin group (16) in 88% yield.24 Subsequent oxidation of the Bpin group of 16 produced the corresponding gem-difluoroalkyl alcohol 17 in 77% yield.25
 | ||
| Scheme 5 Synthetic applications. 6a was used directly without further purification. The yield of 6a was determined by 1H NMR using CH2Br2 as an internal standard. Reaction conditions: (a) LiOtBu, 1,2-DME, 0 °C, 30 h, then 90 °C, 64 h. (b) AgF, THF/D2O/MeOD, 80 °C, 2 h. (c) Cat. Pd(PPh3)4, NaOH, β-bromostyrene, toluene/H2O, 80 °C, 6 h. (d) Cat. Pd(PPh3)4 Cs2CO3, estrone-derived triflate, toluene/H2O, 80 °C, 18 h. (e) Pd/C, H2, MeOH, 60 °C, 3 h. (f) NaOH, H2O2, THF, 0 °C to rt, 2 h. | ||
Conclusions
In summary, we have developed a strategy that enables the synthesis of allyl gem-difluorides through a deborylative allylation/Cope rearrangement between a newly designed 3-boryl-1,1-difluoro allylboronic ester and allyl bromides. Comprehensive experimental and computational studies reveal that the 1,1-difluoro substituents and the boronic ester work cooperatively to lower the barrier of the rearrangement and control the stereochemical outcome. The reaction exhibits broad substrate scope, gram-scale scalability, and versatile synthetic applications, highlighting its utility for constructing fluorinated scaffolds.Author contributions
H. K. and S. C. conceived, designed, and wrote the manuscript. H. K. and S. H. carried out the mechanistic studies. H. K., Y. J., and Y. J. carried out the experiments. S. C. organized the research. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Note added after first publication
This article replaces the version published on 11th December 2025. Table 1 image size and layout have been corrected.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08340k.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) [NRF-2022R1A2C3004731 and RS-2023-00219859]. This research was also supported by the Bio&Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) [RS-2023-00274113]. S. H. Cho thanks Korea Toray Science Foundation for the financial support.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- (a) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (b) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441 RSC; (c) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234 CrossRef CAS.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- (a) J. B. Y. Wang and N. Serradell, Drugs Future, 2006, 31, 788 CrossRef; (b) Y. N. Lamb, Drugs, 2017, 77, 1797 CrossRef CAS PubMed.
- For selected examples of the application of allyl gem-difluorides, see: (a) T. Narumi, K. Tomita, E. Inokuchi, K. Kobayashi, S. Oishi, H. Ohno and N. Fujii, Org. Lett., 2007, 9, 3465 CrossRef CAS PubMed; (b) Y. Yamaki, A. Shigenaga, K. Tomita, T. Narumi, N. Fujii and A. Otaka, J. Org. Chem., 2009, 74, 3272 CrossRef CAS PubMed; (c) M. Bergeron, T. Johnson and J.-F. Paquin, Angew. Chem., Int. Ed., 2011, 50, 11112 CrossRef CAS PubMed; (d) H. Yanai, H. Okada, A. Sato, M. Okada and T. Taguchi, Tetrahedron Lett., 2011, 52, 2997 CrossRef CAS; (e) T. A. Unzner and T. Magauer, Tetrahedron Lett., 2015, 56, 877 CrossRef CAS; (f) L.-n. Tang, G.-y. Liu, J.-h. Li and M. Chen, Org. Lett., 2023, 25, 9064 CrossRef CAS PubMed; (g) G.-y. Liu, L.-n. Tang, J.-h. Li, S. Yang and M. Chen, Chem. Commun., 2024, 60, 4471 RSC.
- D. Tomon and S. Arimitsu, Eur. J. Org Chem., 2025, 28, e202401162 CrossRef CAS.
- For selected examples of the synthesis of allyl gem-difluorides, see: (a) X. Yue, X. Zhang and F.-L. Qing, Org. Lett., 2009, 11, 73 CrossRef CAS PubMed; (b) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (c) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230 CrossRef CAS PubMed; (d) M. Ke, Q. Feng, K. Yang and Q. Song, Org. Chem. Front., 2016, 3, 150 RSC; (e) C.-Q. Wang, Y. Zhang and C. Feng, Angew. Chem., Int. Ed., 2017, 56, 14918 CrossRef CAS PubMed; (f) Z. Zhao, L. Racicot and G. K. Murphy, Angew. Chem., Int. Ed., 2017, 56, 11620 CrossRef CAS PubMed; (g) G. Wu and A. Jacobi von Wangelin, Chem. Sci., 2018, 9, 1795 RSC; (h) W.-H. Guo, H.-Y. Zhao, Z.-J. Luo, S. Zhang and X. Zhang, ACS Catal., 2019, 9, 38 CrossRef CAS; (i) L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701 RSC; (j) L. Liao, R. An, H. Li, Y. Xu, J.-J. Wu and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11010 CrossRef CAS PubMed; (k) F. Ye, Y. Ge, A. Spannenberg, H. Neumann, L.-W. Xu and M. Beller, Nat. Commun., 2021, 12, 3257 CrossRef CAS PubMed; (l) C. Zhu, M.-M. Sun, K. Chen, H. Liu and C. Feng, Angew. Chem., Int. Ed., 2021, 60, 20237 CrossRef CAS PubMed; (m) X.-T. Feng, J.-X. Ren, X. Gao, Q.-Q. Min and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210103 CrossRef CAS PubMed; (n) T. Dong, Y. Ye, Y. Wang, K. P. S. Cheung and G. C. Tsui, Chem.–Asian J., 2023, 18, e202300655 CrossRef CAS PubMed; (o) Z. Fu and G. C. Tsui, Org. Chem. Front., 2024, 11, 4697 RSC; (p) M. Zhou, J.-X. Ren, X.-T. Feng, H.-Y. Zhao, X.-P. Fu, Q.-Q. Min and X. Zhang, Chem. Sci., 2024, 15, 2937 RSC; (q) Z.-X. Wang, N. A. Heckmann, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2025, e202518520 CAS.
- (a) Y.-G. Lou, A.-J. Wang, L. Zhao, L.-F. He, X.-F. Li, C.-Y. He and X. Zhang, Chem. Commun., 2019, 55, 3705 RSC; (b) S. Sakamoto, T. W. Butcher, J. L. Yang and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 60, 25746 CrossRef CAS PubMed.
- For selected examples using 1,1-difluoro allylmetalloids, see: (a) G.-q. Shi and X.-h. Huang, Tetrahedron Lett., 1996, 37, 5401 CrossRef CAS; (b) F. Tellier, M. Baudry and R. Sauvêtre, Tetrahedron Lett., 1997, 38, 5989 CrossRef CAS; (c) P. Veeraraghavan Ramachandran and A. Chatterjee, Org. Lett., 2008, 10, 1195 CrossRef PubMed; (d) P. V. Ramachandran, A. Tafelska-Kaczmarek and A. Chatterjee, J. Org. Chem., 2012, 77, 9329 CrossRef CAS PubMed; (e) Y. Liu, Y. Zhou, Y. Zhao and J. Qu, Org. Lett., 2017, 19, 946 CrossRef CAS PubMed; (f) R. Kojima, S. Akiyama and H. Ito, Angew. Chem., Int. Ed., 2018, 57, 7196 CrossRef CAS PubMed; (g) X. Yang, Z.-H. Cao, Y. Zhou, F. Cheng, Z.-W. Lin, Z. Ou, Y. Yuan and Y.-Y. Huang, Org. Lett., 2018, 20, 2585 CrossRef CAS PubMed; (h) P. H. S. Paioti, J. del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917 CrossRef CAS PubMed; (i) G. Chen, L. Wang, X. Liu and P. Liu, Adv. Synth. Catal., 2020, 362, 2990 CrossRef CAS; (j) N. A. Phillips, G. J. Coates, A. J. P. White and M. R. Crimmin, Chem.–Eur. J., 2020, 26, 5365 CrossRef CAS PubMed; (k) J. Qi, F.-L. Zhang, J.-K. Jin, Q. Zhao, B. Li, L.-X. Liu and Y.-F. Wang, Angew. Chem., Int. Ed., 2020, 59, 12876 CrossRef CAS PubMed; (l) C. Luo, Y. Zhou, H. Chen, T. Wang, Z.-B. Zhang, P. Han and L.-H. Jing, Org. Lett., 2022, 24, 4286 CrossRef CAS PubMed; (m) N. Oyama, S. Akiyama, K. Kubota, T. Imamoto and H. Ito, Eur. J. Org Chem., 2022, 2022, e202200664 CrossRef CAS.
- (a) W. Jo, J. H. Lee and S. H. Cho, Chem. Commun., 2021, 57, 4346 RSC; (b) Y. Lee, S. Han and S. H. Cho, Acc. Chem. Res., 2021, 54, 3917 CrossRef CAS PubMed.
- (a) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415 RSC; (b) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306 CrossRef CAS; (c) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050 RSC; (d) C. Zhang, W. Hu and J. P. Morken, ACS Catal., 2021, 11, 10660 CrossRef CAS PubMed; (e) X. Li, J. Chen and Q. Song, Chem. Commun., 2024, 60, 2462 RSC.
- (a) S. T. Purrington, S. C. Weeks and J. Fluor, Chem, 1992, 56, 165 CAS; (b) E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133 RSC; (c) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923 CrossRef CAS PubMed; (d) Y. Liu, X. Liu and X. Feng, Chem. Sci., 2022, 13, 12290 RSC.
- H. Lee, Y. Lee and S. H. Cho, Org. Lett., 2019, 21, 5912 CrossRef CAS PubMed.
- (a) J. R. Coombs, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 16140 CrossRef CAS PubMed; (b) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581 CrossRef CAS PubMed; (c) W. Sun, L. Wang, Y. Hu, X. Wu, C. Xia and C. Liu, Nat. Commun., 2020, 11, 3113 CrossRef CAS PubMed.
- For selected examples of pericyclic reactions with boron compounds, see: (a) X. Gao and D. G. Hall, J. Am. Chem. Soc., 2003, 125, 9308 CrossRef CAS PubMed; (b) J. Pietruszka and N. Schöne, Angew. Chem., Int. Ed., 2003, 42, 5638 CrossRef CAS PubMed; (c) M. Murakami, S. Ashida and T. Matsuda, J. Am. Chem. Soc., 2004, 126, 10838 CrossRef CAS PubMed; (d) C. Tillin, R. Bigler, R. Calo-Lapido, B. S. L. Collins, A. Noble and V. K. Aggarwal, Synlett, 2019, 30, 449 CrossRef CAS; (e) K. Kanti Das, P. Kumar, D. Ghorai, B. Mondal and S. Panda, Asian J. Org. Chem., 2022, 11, e202100092 CrossRef CAS; (f) Y. Liu, D. Ni and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 18790 CrossRef CAS PubMed; (g) J. M. Posz, N. Sharma, P. A. Royalty, Y. Liu, C. Salome, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2024, 146, 10142 CrossRef CAS PubMed; (h) S. Adak, P. S. Hazra, C. B. Fox and M. K. Brown, Angew. Chem., Int. Ed., 2025, 64, e202416215 CrossRef CAS PubMed; (i) A. Messara, B. Kweon, F. Woge, C. G. Daniliuc, C. Mück-Lichtenfeld and R. Gilmour, J. Am. Chem. Soc., 2025, 147, 36677 CrossRef CAS PubMed; (j) J. Taguchi, Y. Ohata, H. Akimoto, H. Tabuchi, K. Igawa, K. Tomooka, T. Niwa and T. Hosoya, Org. Lett., 2025, 27, 4428 CrossRef CAS PubMed.
- D. V. Vidhani, M. E. Krafft and I. V. Alabugin, J. Am. Chem. Soc., 2016, 138, 2769 CrossRef CAS PubMed.
- (a) W. R. Dolbier, K. S. Medinger, A. Greenberg and J. F. Liebman, Tetrahedron, 1982, 38, 2415 CrossRef CAS; (b) K. A. Black, S. Wilsey and K. N. Houk, J. Am. Chem. Soc., 2003, 125, 6715 CrossRef CAS PubMed.
- S. Wang, H. Wu, X. Tang and G. Huang, Org. Chem. Front., 2024, 11, 2870 RSC.
- L.-G. Zhuo, W. Liao and Z.-X. Yu, Asian J. Org. Chem., 2012, 1, 336 CrossRef CAS.
- For selected examples of the mesomeric effect of fluorine, see: (a) W. A. Sheppard, J. Am. Chem. Soc., 1965, 87, 2410 CrossRef CAS; (b) M. Ohashi, K. Ando, S. Murakami, K. Michigami and S. Ogoshi, J. Am. Chem. Soc., 2023, 145, 23098 CrossRef CAS PubMed.
- Y. Jung and S. H. Cho, Synlett, 2023, 34, 2165 CrossRef CAS.
- S. Wang, J. Zhang, L. Kong, Z. Tan, Y. Bai and G. Zhu, Org. Lett., 2018, 20, 5631 CrossRef CAS PubMed.
- W.-H. Guo, H.-Y. Zhao, Z.-J. Luo, S. Zhang and X. Zhang, ACS Catal., 2019, 9, 38 CrossRef CAS.
- B. Zhang and X. Zhang, Chem. Commun., 2016, 52, 1238 RSC.
- Y. Fan, Z. Huang, Y. Lu, S. Zhu and L. Chu, Angew. Chem., Int. Ed., 2024, 63, e202315974 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2026 |