
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pyridinium cation engineering strategy to achieve high-performance X-ray scintillation of antimony halides
Yuan-Yuan
Zhao
ab,
Jian-Cheng
Chen
ab,
Li
Zhang
c,
Jiabin
Chen
*c,
Qing
Liao
*d,
Zhong-Qiu
Li
 *ac,
Meijin
Lin
cd,
Jiannian
Yao
abc and
Yu-Wu
Zhong
*ac
*ac,
Meijin
Lin
cd,
Jiannian
Yao
abc and
Yu-Wu
Zhong
*ac
aKey Laboratory of Photochemistry, Beijing National Laboratory for Molecular Sciences, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Science, Beijing 100190, China. E-mail: lizhongqiu@iccas.ac.cn; zhongyuwu@fzu.edu.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cInstitute of Molecular Engineering Plus, College of Chemistry, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: jiabinchen@fzu.edu.cn
dCollege of Materials Science and Engineering, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: qingliao@fzu.edu.cn
First published on 8th December 2025
Abstract
While organic–inorganic hybrid metal halides (OIMHs) have emerged as promising X-ray scintillators, a major challenge lies in the precise control of their structural dimensionality and exciton behavior to optimize the scintillation performance. Here, we introduce a pyridinium cation engineering strategy to address this issue. A one-pot reaction of SbCl3 with a pyridine derivative, in the presence of HCl, yields either a 0D or 1D Sb-based OIMH. The 0D structure featuring isolated [SbCl5]2− polyhedra imposes strong quantum confinement, leading to enhanced electron-phonon coupling. This results in an orange-red self-trapped exciton emission at 630 nm, characterized by a large Stokes shift of 262 nm and a near-unity photoluminescence quantum yield. This 0D material demonstrates high-performance X-ray scintillation with a light yield of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 718 photons MeV−1 and a low detection limit of 0.31 µGyair s−1, enabling high-resolution imaging. In contrast, the 1D material consists of corner-sharing inorganic chains. It shows green emission from organic free excitons and poor scintillation performance. This study establishes that pyridinium cation engineering is a potent and promising strategy for the design and optimization of metal halide-based emitters and scintillators.
718 photons MeV−1 and a low detection limit of 0.31 µGyair s−1, enabling high-resolution imaging. In contrast, the 1D material consists of corner-sharing inorganic chains. It shows green emission from organic free excitons and poor scintillation performance. This study establishes that pyridinium cation engineering is a potent and promising strategy for the design and optimization of metal halide-based emitters and scintillators.
Introduction
Scintillators are critical energy transducers that convert high-energy photons like X- or γ-rays into detectable visible light, underpinning technological progress in medical radiography, security screening, and high-energy physics research.1–6 Commercial inorganic scintillators such as NaI:Tl, CsI:Tl, LYSO (Lu2SiO5:Ce), and BGO (Bi4Ge3O12) exhibit high light yields but suffer from some intrinsic limitations.7 For instance, LYSO requires energy-intensive synthesis,8 while CsI:Tl degrades under ambient humidity, compromising their long-term stability.9 Organic scintillators, though solution-processable, face low X-ray absorption coefficients and exciton utilization efficiency, restricting their practical utility.10–12 Organic–inorganic hybrid metal halides (OIMHs), which leverage synergistic coordination between metal ions and organic ligands, have emerged as promising X-ray scintillators due to their solution processability, structural tunability, and exceptional optoelectronic properties.13–15 Among them, lead-based OIHMs have been demonstrated to exhibit high X-ray absorption, a property arising from their heavy elements (Pb, Br/I). Nonetheless, their inherent toxicity and moisture instability impede practical deployment.16,17 Although copper (Cu+) and manganese (Mn2+) alternatives mitigate lead's toxicity concerns, both face some fundamental limitations. Mn2+ emission suffers from spin-forbidden d–d transitions, leading to millisecond-scale lifetimes and narrow spectral tunability (510–650 nm).18,19 Meanwhile, Cu+ aggregates induce localized exciton emission confined to 440–580 nm, which also restricts performance optimization.20 Consequently, there is an urgent and critical need for environmentally robust, high-performance alternatives that feature both scalable fabrication and superior, tunable optoelectronic properties.Recently, antimony-based OIMHs (Sb-OIMHs) have emerged as a promising candidate for high-performance X-ray scintillator. Sb3+'s stereochemical active 5s2 lone pair promotes strong electron-phonon coupling and facilitates self-trapped exciton (STE) formation—a mechanism enabling near-unity photoluminescence quantum yields (PLQYs) and broadband emissions.21–23 Low-dimensional architectures, particularly 0D systems, further exploit quantum confinement to minimize self-absorption losses through large Stokes shifts.24 Most currently reported Sb-OIMHs are based on phosphonium or alkylammonium cations.25–27 For instance, the 0D compound (C19H18P)2Sb2Cl8, built from triphenylmethylphosphonium cations and [Sb2Cl8]2− dimer units, exhibits efficient STE emission with a near-unity PLQY (Fig. 1a).28 This material shows outstanding scintillator performance with a high light yield of 41300 photons MeV−1 and a low detection limit of 45.6 nGyair s−1. The tetrapropylammonium-based 0D material (C12H28N)2SbCl5, consisting of isolated [SbCl5]2− anions, achieves a high PLQY of 96.8%.29 Nevertheless, despite observed radioluminescence (RL) under β-ray excitation, detailed studies on the scintillation properties and X-ray imaging capabilities of ammonium-cation-based antimony halides are scarce. The limited structural diversity offered by these conventional organic moieties restricts systematic tuning of the connectivity of [SbXn] polyhedra and associated exciton dynamics. Moreover, the role of organic components in modulating the emission mechanism (STE vs. free exciton, FE) and their impact on X-ray scintillation performance are still unclear, hindering the rational design of the next-generation Sb-OIMH scintillators.
 | ||
| Fig. 1 (a) Previously reported 0D Sb-OIMHs based on phosphonium or alkylammonium cations. (b) Schematic illustration of the dimensionality control by a pyridinium cation engineering strategy to obtain 0D or 1D Sb-OIMHs with different exciton and RL properties. GS = ground state. ES = excited state. | ||
To address these limitations, we demonstrate here the use of pyridinium cation as a superior alternative to conventional phosphonium or alkylammonium cations for simultaneously regulating the structural dimensionality and exciton behavior of Sb-OIMHs to achieve high-performance X-ray scintillation. Pyridinium derivatives are widely available and possess a great degree of structural tunability. The use of pyridinium derivatives as the organic cations of Sb-OIMHs enable a precise structure and dimensionality control through directional N+–H⋯X hydrogen bonding and Coulombic interactions,30–33 yielding either isolated [SbXn] polyhedra or linked chains. This in turn governs the exciton behavior and emission properties of the resulting materials. In this context, we found that the reaction of two structurally related pyridine derivatives, N-(pyridin-4-yl)benzamide (PBA) and 4-((E)-2-phenylvinyl)pyridine (PVP) in the presence of HCl, with SbCl3 yielded 0D (PBAH+)2SbCl5 and 1D (PVPH+)SbCl4 OIMHs, respectively (Fig. 1b). The 0D architecture of (PBAH+)2SbCl5 promotes intense electron-phonon coupling and efficient STE formation, leading to superior broadband orange-red emission at 630 nm with a large Stokes shift of 262 nm and a near-unity PLQY. In particular, this STE-dominated mechanism delivers exceptional X-ray scintillation performance, achieving a high light yield of 20718 photons MeV−1 and a low detection limit of 0.31 µGyair s−1. In contrast, 1D (PVPH+)SbCl4 shows organic exciton-dominated FE emission and exhibits poor scintillation performance. This work not only provides fundamental insights into how organic cations dictate the structural dimensionality and exciton dynamics of Sb-OIMHs, but also pioneers a pyridinium cation engineering strategy for the development of the next-generation radiation detection materials.
Results and discussion
Structural dimensionality control
The single crystals of (PBAH+)2SbCl5 and (PVPH+)SbCl4 were synthesized via a one-pot reaction procedure using PBA or PVP, SbCl3 and HCl as the starting materials (Fig. 2a; see details in the Experimental section). The experimental power X-ray diffraction (PXRD) patterns of both materials are in good agreement with the simulated patterns derived from the single-crystal X-ray diffraction (SCXRD) data (Fig. 2b and c), confirming the high phase purity and uniformity of the as-synthesized crystals. | ||
| Fig. 2 (a) Schematic of the preparations of (PBAH+)2SbCl5 and (PVPH+)SbCl4 crystals. Inset: photographs of crystals under ambient light and UV irradiation. (b) and (c) Experimental and simulated PXRD patterns of (b) (PBAH+)2SbCl5 and (c) (PVPH+)SbCl4 crystals. (d)–(i) Single-crystal X-ray structures of (d)–(f) (PBAH+)2SbCl5 and (g)–(i) (PVPH+)SbCl4. (e) and (h) Images showing the hydrogen-bonding networks between organic cations and inorganic units. (f) and (i) Images showing the Sb⋯Sb distances among adjacent inorganic units. | ||
SCXRD analyses reveal that (PBAH+)2SbCl5 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , featuring isolated [SbCl5]2− square pyramids with Sb–Cl bond lengths in the range of 2.38–2.77 Å (Fig. S1a). A network of N–H⋯Cl and C
, featuring isolated [SbCl5]2− square pyramids with Sb–Cl bond lengths in the range of 2.38–2.77 Å (Fig. S1a). A network of N–H⋯Cl and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H hydrogen bonds from the protonated pyridinium cation PBA+ (pyridinium N–H⋯Cl: 2.65–2.77 Å; amide N–H⋯Cl: 2.76 Å; amide CO⋯H: 2.47 Å) creates a 0D confinement, which spatially isolates [SbCl5]2− polyhedra and yields extended Sb⋯Sb distances of 6.91–12.82 Å (Fig. 2d–f). The steric and electronic effects of the benzamide group work synergistically with this hydrogen-bonding network to stabilize the overall structure. In contrast, (PVPH+)SbCl4 adopts a monoclinic P21/c structure. The corner-sharing [SbCl4]– polyhedra form 1D zigzag chains along the c-axis (Sb–Cl: 2.39–2.97 Å; Fig. S1b), with a shortest intrachain Sb⋯Sb distances of 4.40 Å (Fig. 2g–i). The PVPH+ cations stabilize the 1D framework via N–H⋯Cl hydrogen bonds (2.55 Å). As discussed below, the structural differences of these two materials lead to distinct emission and radioluminescence properties. In particular, the extended Sb⋯Sb separation (>6.9 Å) in 0D systems enables unconstrained lattice distortion, while 1D chains enforce rigid polyhedral connectivity that suppresses structural flexibility.
O⋯H hydrogen bonds from the protonated pyridinium cation PBA+ (pyridinium N–H⋯Cl: 2.65–2.77 Å; amide N–H⋯Cl: 2.76 Å; amide CO⋯H: 2.47 Å) creates a 0D confinement, which spatially isolates [SbCl5]2− polyhedra and yields extended Sb⋯Sb distances of 6.91–12.82 Å (Fig. 2d–f). The steric and electronic effects of the benzamide group work synergistically with this hydrogen-bonding network to stabilize the overall structure. In contrast, (PVPH+)SbCl4 adopts a monoclinic P21/c structure. The corner-sharing [SbCl4]– polyhedra form 1D zigzag chains along the c-axis (Sb–Cl: 2.39–2.97 Å; Fig. S1b), with a shortest intrachain Sb⋯Sb distances of 4.40 Å (Fig. 2g–i). The PVPH+ cations stabilize the 1D framework via N–H⋯Cl hydrogen bonds (2.55 Å). As discussed below, the structural differences of these two materials lead to distinct emission and radioluminescence properties. In particular, the extended Sb⋯Sb separation (>6.9 Å) in 0D systems enables unconstrained lattice distortion, while 1D chains enforce rigid polyhedral connectivity that suppresses structural flexibility.
The chemical states of the constituent elements of (PBAH+)2SbCl5 and (PVPH+)SbCl4 were further verified by X-ray photoelectron spectroscopy (XPS) studies (Fig. S2). Furthermore, these materials demonstrate good thermal and environmental stability. Thermogravimetric analysis (TGA) indicates negligible weight loss for both compounds below 200 °C (Fig. S3). Additionally, their PXRD patterns remain unchanged after six months of exposure to ambient atmospheric conditions (Fig. S4).
Exciton dynamics
Scanning electron microscopy (SEM) shows that (PBAH+)2SbCl5 forms as isolated rhombic blocks with widths of 50–80 µm (Fig. 3a), while (PVPH+)SbCl4 grows as elongated rods with lengths of a few hundreds of µm and widths of 15–25 µm (Fig. 3b). Under UV excitation, the 0D (PBAH+)2SbCl5 crystals emit intense orange-red light, whereas the 1D (PVPH+)SbCl4 crystals show green emission. This dichotomy is consistent with their different bandgap energies (3.10 and 2.58 eV), respectively; Fig. S5. | ||
| Fig. 3 (a) and (b) SEM images of (a) (PBAH+)2SbCl5 and (b) (PVPH+)SbCl4. Inset: corresponding fluorescence microscopy images under UV excitation. (c) Time-resolved PL decay profiles of two crystals. (d) and (e) PLE and PL spectra of (d) (PBAH+)2SbCl5 and (e) (PVPH+)SbCl4. (f) Temperature-dependent emission contour maps of (PBAH+)2SbCl5. (g) Normalized temperature-dependent PL spectra of two crystals. (h) Plots of FWHM versus temperature. (i) Plots of PL intensity versus reciprocal temperature. | ||
The 0D (PBAH+)2SbCl5 crystals exhibit hallmark signatures of STE emission with a broad and featureless photoluminescence (PL) band centered at 630 nm with a substantial Stokes shift of 262 nm and full width at half maxima (FWHM) of 135 nm (Fig. 3d). This emission possesses a microsecond-scale lifetime (τ = 4.42 µs, Fig. 3c) and a near-unity PLQY (>99%; Fig. S6). Photoluminescence excitation (PLE) spectral analysis suggests that STE-dominated emission is the exclusive radiative pathway of (PBAH+)2SbCl5 (Fig. S7). The 0D structure with a geometric isolation of the inorganic units gives rise to a quantum-confined environment that intensifies electron-phonon coupling and thereby enables efficient STE formation.34,35 The strength of this coupling is evidenced by a high Huang–Rhys factor (S = 12.45, see below). This coupling triggers significant Jahn–Teller distortion upon photoexcitation. The resultant lattice deformation creates localized potential wells that trap excitons, facilitating efficient triplet-to-singlet radiative recombination (3P1 → 1S0) at Sb3+ centers.36 The broadband spectrum, large Stokes shift, and microsecond-scale lifetime of the STE-dominated emission offers distinct advantages over other processes like phosphorescence or thermally delayed fluorescence. These properties are particularly beneficial for X-ray scintillation, as they effectively suppress self-absorption losses and enable efficient photon integration with minimal afterglow, which is crucial for achieving high-resolution imaging and low-dose detection.
In contrast, the 1D (PVPH+)SbCl4 system displays fundamentally distinct exciton dynamics. It shows narrow green emission at 515 nm with FWHM of 86 nm, nanosecond lifetime of 3.11 ns (Fig. 3c), minimal Stokes shift of 45 nm (Fig. 3e), and moderate PLQY of 27.4% (Fig. S8). These characteristics unequivocally excludes STE emission.37,38 The PL and PLE spectral profiles are independent of the excitation and emission conditions (Fig. S9), suggesting a single emission center. Control experiments show that the PVP·HCl organic salt has similar photophysics with emission maximum (λem) at 463 nm, PLQY of 31.77%, lifetime of 7.33 ns, and FWHM of 83 nm (Fig. S10 and S11). This suggests that the emission of (PVPH+)SbCl4 is very likely of the organic origin. The emission redshift of (PVPH+)SbCl4versus PVP·HCl is caused by the influence of the inorganic units. The compressed 1D geometry induces strong lone-pair repulsion among adjacent Sb3+ centers, quenching Sb-centered emission and funneling excitonic energy to organic cations (PVPH+).39 The key photophysical parameters of both materials are summarized in Table S6, and their emissions are plotted using the Commission Internationale de l’Eclairage (CIE) chromaticity coordinates in Fig. S12. After storing for six months under ambient conditions, the PLQY values decrease from near-unity to 74.75% for (PBAH+)2SbCl5 and from 27.4% to 9.84% for (PVPH+)SbCl4, respectively; while their emission profiles remain essentially unchanged (Fig. S13 and S14). The PLQY reduction is likely initiated by surface hydrolysis due to the hygroscopic pyridinium cations, leading to the generation of non-radiative surface defects. The unchanged emission spectra suggest that this is a surface-dominated process, while the bulk emission centers remain stable.
To elucidate the exciton–phonon interaction mechanism, temperature-dependent PL spectra of the above two crystals have been investigated (Fig. 3f–i and S15). Both compounds exhibit a continuous blue-shift of their emission bands upon heating (Δλ ≈ 10–15 nm from 80–300 K, Fig. 3g), accompanied by progressive peak broadening. This phenomenon arises from two competing effects: thermally-induced lattice expansion, which reduces crystal field splitting, and enhanced electron-phonon coupling, which promotes exciton delocalization. Both are characteristic behaviors in Sb3+-based halides.40 Concurrently, the emission intensities decrease exponentially with temperature due to the thermal activation of non-radiative pathways (Fig. S15). The observed linewidth broadening reflects a strengthening of exciton–lattice interactions.
Quantitative assessment of the electron-phonon coupling strength is achieved through the Huang–Rhys factor (S) analysis using the temperature-dependent FWHM evolution modeled by eqn (1):
 | (1) |
The exciton binding energies (Eb) are further extracted from the Arrhenius analysis of integrated PL intensity by eqn (2):
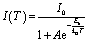 | (2) |
Theoretical calculations
Density functional theory (DFT) calculations provide fundamental insights into the electronic origins of the contrasting emission mechanisms between 0D and 1D architectures. Band structure analysis reveals that (PBAH+)2SbCl5 adopts an indirect bandgap with a bandgap energy Eg of 2.02 eV (Fig. 4a), whereas (PVPH+)SbCl4 exhibits a direct bandgap with Eg of 1.72 eV (Fig. 4d). While these values underestimate experimental optical gaps (3.10 eV vs. 2.58 eV),45,46 the relative trend aligns with the spectroscopic measurements. Crucially, the projected density of states (PDOS) unravels the atomic-scale dichotomy. In 0D (PBAH+)2SbCl5, the valence band maximum (VBM) arises from the hybridization of Sb 5s/5p and Cl 3p orbital, while the conduction band minimum (CBM) is dominated by the π* orbitals of PBA+ cations (Fig. 4b). This inorganic VBM and organic CBM character creates a hybrid charge-transfer state that localizes excitons on the [SbCl5]2− clusters prior to the recombination through organic manifolds, directly enabling STE formation.47 This theoretical model is corroborated by the UV-vis absorption data (Fig. S16), which shows a distinct low-energy shoulder band at 360–420 nm. This feature is attributed to a charge-transfer transition between the inorganic [SbCl5]2− clusters and the organic PBAH+ cations, as it cannot be assigned to the intrinsic transitions of either isolated component. In contrast, in 1D (PVPH+)SbCl4, both VBM and CBM originate from the π and π* orbitals of PVP+ (C 2p/N 2p, Fig. 4e), signifying purely organic excitons confined within the cation framework.48 This dimensional divergence is further visualized in the charge density difference maps (Fig. 4c and f). The excitation of 0D system triggers pronounced electron density redistribution from Sb–Cl bonds to pyridinium group, leading to exciton localization across inorganic–organic interfaces. In contrast, the excitation of 1D analogue displays intramolecular polarization within the PVP+ moiety, consistent with the localized π → π* transitions. | ||
| Fig. 4 (a) and (d) Calculated band structures, (b) and (e) density of states (DOS), and (c) and (f) electron density distributions of VBM and CBM of (a)–(c) (PBAH+)2SbCl5 and (d)–(f) (PVPH+)SbCl4. | ||
X-ray scintillation and imaging
Theoretical X-ray attenuation analysis reveals that both Sb-OIMHs exhibit reduced absorption coefficients relative to the inorganic scintillator BGO across photon energies of 1–1000 keV using the XCOM web database, consistent with their lower crystalline densities (Fig. 5a).49 Under X-ray excitation, both materials exhibit RL spectra very similar to their PL profiles, confirming the preservation of the same emission centers and recombination pathways regardless of the excitation source (Fig. 5b). The slight redshift (∼5 nm) of the observed RL versus corresponding PL spectra of both samples likely originates from the instrumental calibration factors. The relative light yield of (PBAH+)2SbCl5 was calculated to be 20718 photons MeV−1 according to the commercial BGO scintillator as a reference. This performance is 41-fold higher than the 1D (PVPH+)SbCl4 counterpart (505 photons MeV−1; Fig. 5b and S17). The excellent RL performance of the 0D (PBAH+)2SbCl5 originates from the synergistic advantages of STE emission including the near-unity radiative efficiency, a high exciton binding energy that suppresses thermal quenching, and a large Stokes shift that eliminate self-reabsorption.50 In contrast, the organic exciton emission of the 1D (PVPH+)SbCl4 suffers from competitive non-radiative decay and inherent self-absorption, fundamentally limiting its scintillation output.
 | ||
| Fig. 5 (a) X-ray attenuation coefficient diagram of (PBAH+)2SbCl5, (PVPH+)SbCl4 and BGO (from the NIST XCOM database). (b) RL spectra measured under X-ray irradiation (50 kV, 79 µA, dose rate = 278 µGyair s−1). (c) The functional correlation between RL intensity and dose rate (0.7–278 µGyair s−1) of X-ray for (PBAH+)2SbCl5. (d) Radiation resistance test of (PBAH+)2SbCl5 under prolonged X-ray exposure (278 µGyair s−1 for 7000 s). (e) X-ray images of the line-pair card (type 25N: 3.5–20 lp per mm) and (f) corresponding pixel intensity profile across pixel positions (resolution: 8.0 lp per mm, MTF = 0.21 ± 0.01) under X-ray exposure. (g)–(i) Photographs captured under white light and X-ray illumination with (PBAH+)2SbCl5 of (g) a capsule with a metallic, (h) a dried fish, (i) a printed circuit board. | ||
Further evaluation establishes that (PBAH+)2SbCl5 is a premier low-dose detection platform. It exhibits an outstanding dose linearity across the clinically relevant ranges of 0.7–278 µGyair s−1 (Fig. 5c, S18 and Table S7), with a rather low detection limit of 0.31 µGyair s−1 at a signal-to-noise ratio (SNR) of 3. This represents 94% reduction versus the standard medical diagnostics (5.5 µGyair s−1).51 Radiation hardness tests confirm the material's exceptional stability, showing <1% signal attenuation after prolonged exposure for 7000 s at 278 µGyair s−1 and negligible degradation through 20 operational cycles (1200 s total duration; Fig. 5d and S19). These metrics surpass most conventional scintillators in both sensitivity and operational stability, attributed to the material's robust STE potential wells and high crystallinity. Critically, a comparative analysis (Table S8) demonstrates that this 0D (PBAH+)2SbCl5 exhibits comparable or even superior overall scintillation performance to other reported Sb-based OIHMs, making it a valuable alternative candidate for scintillation applications.
The translation of the superior scintillation properties of (PBAH+)2SbCl5 into practical applications has been demonstrated through X-ray imaging studies (Fig. S20). The solution-processed scintillator screens, fabricated via 24 hours ball milling of (PBAH+)2SbCl5 crystals in dichloromethane followed by vacuum filtration, achieves a remarkable spatial resolution of 8.0 lp per mm (MTF = 0.21) as quantified through standardized line-pair tests (Fig. 5e and f).52–54 This excellent X-ray imaging performance enables clear visualization of sub-millimeter mechanical features in encapsulated spring assemblies, trabecular bone microarchitecture in biological specimens, and micron-scale circuit traces in multilayer PCBs (Fig. 5g–i). The high contrast resolution and low noise characteristics observed across these diverse imaging targets confirm the material's versatility for both medical radiography and industrial inspection applications.
Conclusions
In summary, we demonstrate a pyridinium cation engineering method to control the structural dimensionality and exciton behavior of Sb-OIMH scintillators. By introducing an amide linker as an additional hydrogen-bonding site, a 0D Sb-OIMH with isolated [SbCl5]2− polyhedra has been obtained, which exhibit strong electron-phonon coupling and efficient STE emission. This scintillator is characterized with a high light yield and low detection limit, making it suitable for high-resolution X-ray imaging. In contrast, a 1D Sb-OIMH with compressed Sb⋯Sb distances has been obtained with 4-((E)-2-phenylvinyl)pyridine, which favors the emission of organic free excitons but exhibits poor scintillation. This work establishes that a delicate change of the pyridinium cation may have a significant impact on the dimensionality, exciton property, and scintillation performance of the resulting OIMH. Pyridinium cation engineering is believed to become a promising strategy for designing and optimizing of metal halide emitters and scintillators. Our ongoing research efforts focus on the application of this pyridinium cation engineering strategy to other lead-free metal halide systems. We anticipate this approach will enable the rational design of metal halides with tailored optoelectronic properties.Author contributions
Conceptualization: Y.-W. Zhong, Z.-Q. Li, and J. Yao. Methodology: Y.-W. Zhong, Z.-Q. Li, Q. Liao, and J. Chen. Investigation: Y.-Y. Zhao, L. Zhang, Q. Liao, and J. Chen. Visualization: Y.-Y. Zhao, Z.-Q. Li, Y.-W. Zhong, and J.-C. Chen. Funding acquisition: Z.-Q. Li, Q. Liao, and Y.-W. Zhong. Project administration: Z.-Q. Li, Y.-W. Zhong, Q. Liao and J. Chen. Supervision: Y.-W. Zhong, J. Yao, and M. Lin. Writing – original draft: Y.-Y. Zhao. Writing – review & editing: Y.-W. Zhong, Z.-Q. Li, and Q. Liao.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of experimental methods, characterisation data. See DOI: https://doi.org/10.1039/d5sc08289g.CCDC 2363201 and 2431220 contain the supplementary crystallographic data for this paper.55a,b
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grants 22305251 and 52303222) and National Key R&D Program of China (2022YFA1204401 and 2023YFE0125200). Z.-Q. L. acknowledges the support of Beijing Molecular Science (BMS) Junior Fellow of BNLMS.Notes and references
- W. Ma, Y. Su, Q. Zhang, C. Deng, L. Pasquali, W. Zhu, Y. Tian, P. Ran, Z. Chen, G. Yang, G. Liang, T. Liu, H. Zhu, P. Huang, H. Zhong, K. Wang, S. Peng, J. Xia, H. Liu, X. Liu and Y. M. Yang, Nat. Mater., 2021, 21, 210–216 CrossRef PubMed.
- J.-X. Wang, L. Gutiérrez-Arzaluz, X. Wang, T. He, Y. Zhang, M. Eddaoudi, O. M. Bakr and O. F. Mohammed, Nat. Photonics, 2022, 16, 869–875 CrossRef CAS.
- J. Fan, H. Li, W. Liu and G. Ouyang, Angew. Chem., Int. Ed., 2025, 64, e202425661 CrossRef CAS PubMed.
- Y. Chen, S. Niu, Y. Li, W. Dou, X. Yang, C. Shan and G. Shen, Adv. Mater., 2024, 36, 2404656 CrossRef CAS PubMed.
- X. Yan, S.-Y. Song, S. Liang, K.-K. Liu, X.-T. Liu and C. Lu, Chem. Sci., 2025, 16, 16770–16779 RSC.
- H. Chen, X. Yang, Y. Ye, H. Hong, Q. Wei, Y. Zhu and M. J. Lin, Angew. Chem., Int. Ed., 2025, 64, e202506118 CrossRef CAS.
- P. Singh, G. Dosovitskiy and Y. Bekenstein, ACS Nano, 2024, 18, 14029–14049 CrossRef CAS PubMed.
- L. Fan, Y. Wu, Z. Zhang, Y. Shi and J. Xie, Opt. Mater., 2020, 99, 109563 CrossRef CAS.
- Z. Wang, R. T. Williams, J. Q. Grim, F. Gao and S. Kerisit, Phys. Status Solidi B, 2013, 250, 1532–1540 CrossRef CAS.
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang, Z. Zhao, M. Singh, C. Lin, H. Wang, W. Jia, Q. Wang, J. Zhi, C. Dong, X. Jiang, Y. Tang, X. Xie, Y. Yang, J. Wang, Q. Chen, Y. Wang, H. Yang, G. Zhang, Z. An, X. Liu and W. Huang, Nat. Photonics, 2021, 15, 187–192 CrossRef CAS.
- Y.-H. Chen, G.-Z. Zhang, F.-H. Chen, S.-Q. Zhang, X. Fang, H.-M. Chen and M.-J. Lin, Chem. Sci., 2024, 15, 7659–7666 RSC.
- Y. Zhang, M. Chen, X. Wang, M. Lin, H. Wang, W. Li, F. Chen, Q. Liao, H. Chen, Q. Chen, M. Lin and H. Yang, CCS Chem., 2024, 6, 334–341 CrossRef CAS.
- W. Wei, Y. Zhang, Q. Xu, H. Wei, Y. Fang, Q. Wang, Y. Deng, T. Li, A. Gruverman, L. Cao and J. Huang, Nat. Photonics, 2017, 11, 315–321 CrossRef CAS.
- J.-H. Chen, J.-B. Luo, Z.-L. He, Q.-P. Peng, J.-H. Wei, Z.-Z. Zhang, X.-X. Guo and D.-B. Kuang, Chem. Sci., 2025, 16, 9375–9384 RSC.
- Q. Zhou, W. Li, J. Xiao, A. Li and X. Han, Adv. Funct. Mater., 2024, 34, 2402902 CrossRef CAS.
- H. Wei, Y. Fang, P. Mulligan, W. Chuirazzi, H.-H. Fang, C. Wang, B. R. Ecker, Y. Gao, M. A. Loi, L. Cao and J. Huang, Nat. Photonics, 2016, 10, 333–339 CrossRef CAS.
- A. Xie, F. Maddalena, M. E. Witkowski, M. Makowski, B. Mahler, W. Drozdowski, S. V. Springham, P. Coquet, C. Dujardin, M. D. Birowosuto and C. Dang, Chem. Mater., 2020, 32, 8530–8539 CrossRef CAS.
- C. Wang, Y. Li and Z. Deng, Chem. Sci., 2025, 16, 15796–15814 RSC.
- J.-Y. Yao, Q.-R. Ding, H. Zeng, J.-Y. Wang, C.-M. Shi, L.-J. Xu and Z.-N. Chen, Angew. Chem., Int. Ed., 2025, 64, e202502099 CrossRef CAS PubMed.
- H. Zhang, B. Yu, Y. Tan, A. Ying, W. He, Z. Jia, Q. Lin and S. Gong, Sci. China:Chem., 2025, 68, 4245–4254 CrossRef CAS.
- W. Han, P. Cheng, J. Guan, Q. Li, Y. Wang, Z. Wang, T. Rasing, Y. Zheng, J. Xu and X. H. Bu, Angew. Chem., Int. Ed., 2025, 64, e202500786 CrossRef CAS PubMed.
- H. Liu, T. B. Shonde, F. Gonzalez, O. J. Olasupo, S. Lee, D. Luong, X. Lin, J. S. R. Vellore Winfred, E. Lochner, I. Fatima, K. Hanson and B. Ma, Adv. Mater., 2023, 35, 2209417 CrossRef CAS PubMed.
- D.-Y. Li, H.-Y. Kang, Y.-H. Liu, J. Zhang, C.-Y. Yue, D. Yan and X.-W. Lei, Chem. Sci., 2024, 15, 953–963 RSC.
- T. Huang and B. Zou, Nanomaterials, 2023, 13, 2867 CrossRef CAS PubMed.
- Z. Xu, N. Li, X. Yan, X. Wang, T. He, Z. Yang and S. Liu, Adv. Opt. Mater., 2024, 12, 2301477 CrossRef CAS.
- H. Huang, Y. Yang, S. Qiao, X. Wu, Z. Chen, Y. Chao, K. Yang, W. Guo, Z. Luo, X. Song, Q. Chen, C. Yang, Y. Yu and Z. Zou, Adv. Funct. Mater., 2024, 34, 2309112 CrossRef CAS.
- H. Chen, H. Cui, T. Jiang, Z. Feng, Y. Yan, S. Liu and Q. Zhao, J. Mater. Chem. C, 2024, 12, 12325–12331 RSC.
- H. Meng, Y. Li, F. Zhang, S. Niu, M. Zhu, Z. Shi and G. Shen, Adv. Funct. Mater., 2025, 35, 2412597 CrossRef CAS.
- L. Lian, P. Zhang, X. Zhang, Q. Ye, W. Qi, L. Zhao, J. Gao, D. Zhang and J. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 58908–58915 CrossRef CAS.
- Z.-Q. Li, D.-X. Ma, F.-F. Xu, T.-X. Dan, Z.-L. Gong, J.-Y. Shao, Y.-S. Zhao, J. Yao and Y.-W. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202205033 CrossRef CAS PubMed.
- Y.-Y. Zhao, Z.-Q. Li, J.-Y. Shao and Y.-W. Zhong, Cryst. Growth Des., 2023, 23, 9086–9093 CrossRef CAS.
- L. Gao, G. V. Baryshnikov, A. Ali, A. Kuklin, C. Qian, X. Zhang, F. Chen, T. Yi and H. Wu, Angew. Chem., Int. Ed., 2024, 63, e202318497 CrossRef CAS PubMed.
- S. Xiong, Y. Xiong, D. Wang, Y. Pan, K. Chen, Z. Zhao, D. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2301874 CrossRef CAS PubMed.
- J.-F. Liao, Z. Zhang, L. Zhou, Z. Tang and G. Xing, Angew. Chem., Int. Ed., 2024, 63, e202404100 CrossRef CAS PubMed.
- Z. Li, Y. Li, P. Liang, T. Zhou, L. Wang and R.-J. Xie, Chem. Mater., 2019, 31, 9363–9371 CrossRef CAS.
- K. M. McCall, V. Morad, B. M. Benin and M. V. Kovalenko, ACS Mater. Lett., 2020, 2, 1218–1232 CrossRef CAS PubMed.
- R. Chen, C. Sun, X. Cheng, Y. Lin, J. Zhou, J. Yin, B.-B. Cui and L. Mao, Inorg. Chem., 2023, 62, 9722–9731 CrossRef CAS.
- H. Peng, S. Yu, Q. Wei, L. Kong, Z. Du, J. Zhao and B. Zou, Adv. Funct. Mater., 2024, 34, 2411807 CrossRef CAS.
- C. Sun, Z. Deng, Z. Li, Z. Chen, X. Zhang, J. Chen, H. Lu, P. Canepa, R. Chen and L. Mao, Angew. Chem., Int. Ed., 2023, 62, e202216720 CrossRef CAS PubMed.
- D.-Y. Li, J.-H. Song, Z.-Y. Xu, Y.-J. Gao, X. Yin, Y.-H. Hou, L.-J. Feng, C.-Y. Yue, H. Fei and X.-W. Lei, Chem. Mater., 2022, 34, 6985–6995 CrossRef CAS.
- Z. Ma, X. Ji, S. Lin, X. Chen, D. Wu, X. Li, Y. Zhang, C. Shan, Z. Shi and X. Fang, Adv. Mater., 2023, 35, 2300731 CrossRef CAS PubMed.
- Y. Hui, S. Chen, R. Lin, W. Zheng and F. Huang, Mater. Chem. Front., 2021, 5, 7088–7107 RSC.
- X. Han, P. Cheng, S. Han, Z. Wang, J. Guan, W. Han, R. Shi, S. Chen, Y. Zheng, J. Xu and X.-H. Bu, Chem. Sci., 2024, 15, 3530–3538 RSC.
- C. Li, Z. Luo, Y. Liu, Y. Wei, X. He, Z. Chen, L. Zhang, Y. Chen, W. Wang, Y. Liu, X. Chang and Z. Quan, Adv. Opt. Mater., 2022, 10, 2102746 CrossRef CAS.
- D. Chen, S. Hao, G. Zhou, C. Deng, Q. Liu, S. Ma, C. Wolverton, J. Zhao and Z. Xia, Inorg. Chem., 2019, 58, 15602–15609 CrossRef CAS PubMed.
- Q. Mo, Q. Qian, Y. Shi, W. Cai, S. Zhao and Z. Zang, Adv. Opt. Mater., 2022, 10, 2201509 CrossRef CAS.
- H. Meng, B. Chen, W. Zhu, Z. Zhou, T. Jiang, X. Xu, S. Liu and Q. Zhao, Laser Photonics Rev., 2023, 17, 2201007 CrossRef CAS.
- B. Zhuang, Z. Jin, J. Deng, H. Xiong, W. Li and J. Fan, Adv. Opt. Mater., 2023, 11, 2300888 CrossRef CAS.
- S.-B. Xiao, X. Zhang, X. Mao, H.-J. Yang, Z.-N. Chen and L.-J. Xu, Adv. Funct. Mater., 2024, 34, 2404003 CrossRef CAS.
- J. Perego, C. X. Bezuidenhout, I. Villa, F. Cova, R. Crapanzano, I. Frank, F. Pagano, N. Kratochwill, E. Auffray, S. Bracco, A. Vedda, C. Dujardin, P. E. Sozzani, F. Meinardi, A. Comotti and A. Monguzzi, Nat. Commun., 2022, 13, 3504 CrossRef CAS PubMed.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang, Z. Yi, J. Li, X. Xie, Y. Wang, Y. Li, D. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. Yang, W. Huang and X. Liu, Nature, 2018, 561, 88–93 CrossRef CAS PubMed.
- K. Masaoka, Opt. Express, 2019, 27, 1345 CrossRef.
- R. Gerst, Z. Cseresnyés and M. T. Figge, Nat. Methods, 2023, 20, 168–169 CrossRef CAS PubMed.
- L.-J. Xu, X. Lin, Q. He, M. Worku and B. Ma, Nat. Commun., 2020, 11, 4329 CrossRef CAS PubMed.
- (a) CCDC 2363201: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2kb39b; (b) CCDC 2431220: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mlwgl.
| This journal is © The Royal Society of Chemistry 2026 |