
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed sequential distal C–H alkenylation and π-allylic amination of arylacetic acids using MBH acetates: access to macrocyclic lactams
Perumal
Muthuraja
,
Prabhat Kumar
Maharana†
 ,
Tamilthendral
Veerappan†
,
Subhradeep
Kar
and
Tharmalingam
Punniyamurthy
*
,
Tamilthendral
Veerappan†
,
Subhradeep
Kar
and
Tharmalingam
Punniyamurthy
*
Department of Chemistry, Indian Institute Technology Guwahati, Guwahati 781039, India. E-mail: tpunni@iitg.ac.in
First published on 10th November 2025
Abstract
Palladium(II)-catalyzed directed meta-selective C–H functionalization of arylacetic acids has been accomplished utilizing Morita–Baylis–Hillman (MBH) acetates as the coupling partner to furnish β-aryl MBH acetates that can be converted into 14-membered macrocyclic lactams employing water as the oxygen source via a π-allyl intermediate. The sequential meta/meta′ C–H difunctionalization can be accomplished with varied coupling partners. Mechanistic investigations underscore the roles of the nitrile-directing template, palladium(II)-catalysis and ligand in meta-selectivity. The macrocyclization pathway has been validated through an 18O-labeling experiment and the photophysical studies reveal distinct fluorescence in the selected macrocycles. In addition, the selected lactams exhibited biocompatibility with Vero cells and dose-dependent cytotoxicity against MCF-7 cells, highlighting their therapeutic potential. The substrate scope, functional group tolerance, selectivity, photophysical and biological properties, and the late-stage functionalization of drug molecules as well as natural product derivatives are important practical features.
Introduction
Transition-metal-catalyzed carbon–carbon bond formation via direct C–H functionalization has emerged as a powerful synthetic tool in modern organic chemistry,1 enabling efficient and straightforward construction of molecular architectures relevant to natural products, pharmaceuticals and advanced materials.2 In this context, owing to the ubiquity of C–H bonds in organic molecules, achieving site-selective functionalization is a challenging goal.3 Thus, there has been a significant surge of interest towards developing effective synthetic strategies to functionalize the unreactive C–H bonds.4 While the directing group (DG) approach has delivered significant outcomes for reactions at proximal C–H bonds,5 distal functionalization remains a long-standing goal due to thermodynamic instability of the comparatively fluxional metallacycle intermediates.6 Contextually, subsequent modifications in the geometry as well as the structure and electronic nature of DGs have resulted in several successful distal functionalizations.7 However, owing to the significant reliance of these approaches on the rigidity of the transition-state, the template-based pathways are mostly confined to relatively shorter chain aryl-alkanoic acids wherein the distal C–H bond is proximal and accessible.8 In contrast, in the case of substrates with longer alkyl chains, the DG dangles more freely, thereby making it entropically challenging to carry out the activation and functionalization of the C–H bond at a remote site. Interestingly, nitrile-based templates have opened new avenues in distal C–H bond functionalization,9 and showed promising potential in the synthesis of macrocyclic scaffolds, offering advantages in terms of step and atom economy, highlighting their growing relevance in synthetic applications.10 However, while previous reports depict the utilization of these templates solely as DGs, which are removed subsequently after the C–H functionalization, their use as a functional handle in achieving macrocyclization remains unexplored. Considering the widespread presence of the arylacetic acid motif in therapeutics such as ibuprofen, ketoprofen, oxybutynin and glycopyrrolate,11 their utilization to access the less explored macrocyclic chemical space would be valuable. Moreover, C–H functionalization with MBH adducts offers an ideal platform, allowing the conversion of simple and inexpensive building blocks into structurally diverse frameworks. Although, they have been known for their role in ortho-selective functionalization,12 their utilization in distal functionalization is yet to be studied (Fig. 1a). Utilizing MBH adducts for achieving remote alkenylation and exploiting the tethered acetate towards achieving late-stage lactamization would be of wider interest.13 Recently, the Yu group elegantly showed a Pd-catalyzed meta-selective C–H functionalization using an indolyl template for macrocyclization (Fig. 1b),10 while Yang and co-workers reported a Rh-catalyzed macrocyclization employing the ortho-selective C–H functionalization of arenes with MBH adducts (Fig. 1c).12e This is particularly attractive as they play a crucial role in drug discovery with natural product-based fragments contributing to the progress of therapeutics such as largazole, geldanamycin, epothioline B and Ixabepilone (Fig. 1d).14 Herein, we report a Pd(II)-catalyzed meta-selective C–H functionalization of α-hydroxy arylacetic acids with MBH acetates to afford β-aryl MBH acetates that can be cyclized employing Pd(II)-catalysis to furnish 14-membered lactams. The procedure can be extended for meta/meta′ C–H difunctionalization with diverse coupling partners (Fig. 1e). The selectivity, substrate scope, functional group tolerance, application to access macrocyclic lactams, difunctionalization and synthetic potential are important practical features. | ||
| Fig. 1 Pharmaceutical importance of macrocyclic compounds and development of synthetic strategies for macrocyclization. | ||
Results and discussion
We began the optimization studies utilizing α-hydroxy arylacetic acid (mandelic acid, 1a) with MBH acetate 2a as the model substrates (Tables 1 and S1–S5, SI). To our delight, the meta-selective C–H alkenylation occurred to furnish β-aryl MBH acetate 3a (template T1) in 79% yield with 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z-selectivity as the sole product without affecting the ortho-C–H bond, when the substrates 1a and 2a were stirred with Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (60 mol%) and AgOAc (1 equiv.) at 90 °C for 18 h in HFIP. Among the templates screened, T1 exhibited the best results, while T4 and T5 produced 56% and 59% yields, respectively (Table S1, SI). In contrast, template T2 without the nitrile DG was an unsuccessful substrate. A similar result was observed with T3 having the nitrile DG at the third position of the aryl ring. The use of Pd(TFA)2 in place of Pd(OAc)2 led to a drop in the yield to 35% (Entry 2). Of the amino acid ligands studied, N-Ac-Gly-OH, N-Ac-L-Leu-OH and Fmoc-Gly-OH, the former yielded the best outcome (Entries 3 and 4). The reaction utilizing AgOAc was superior to that of Ag2CO3 and AgTFA (entries 5 and 6). HFIP was the solvent of choice, whereas (CH2Cl)2 and TFE gave inferior results (Entries 7 and 8). Control experiments confirmed that the combination of the Pd(II)-catalyst, template and ligand was crucial to achieve selective functionalization.
:1 E/Z-selectivity as the sole product without affecting the ortho-C–H bond, when the substrates 1a and 2a were stirred with Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (60 mol%) and AgOAc (1 equiv.) at 90 °C for 18 h in HFIP. Among the templates screened, T1 exhibited the best results, while T4 and T5 produced 56% and 59% yields, respectively (Table S1, SI). In contrast, template T2 without the nitrile DG was an unsuccessful substrate. A similar result was observed with T3 having the nitrile DG at the third position of the aryl ring. The use of Pd(TFA)2 in place of Pd(OAc)2 led to a drop in the yield to 35% (Entry 2). Of the amino acid ligands studied, N-Ac-Gly-OH, N-Ac-L-Leu-OH and Fmoc-Gly-OH, the former yielded the best outcome (Entries 3 and 4). The reaction utilizing AgOAc was superior to that of Ag2CO3 and AgTFA (entries 5 and 6). HFIP was the solvent of choice, whereas (CH2Cl)2 and TFE gave inferior results (Entries 7 and 8). Control experiments confirmed that the combination of the Pd(II)-catalyst, template and ligand was crucial to achieve selective functionalization.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%)b (E/Z)c |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol), Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (60 mol%), AgOAc (0.1 mmol), HFIP (1 mL), 90 °C, 18 h, air, pressure tube. b Isolated yield. c Determined by 400 MHz 1H NMR. HFIP = 1,1,1,3,3,3-hexafluoroisopropanol. TFE = 2,2,2-trifluoroethanol. | ||
| 1 | None | 79 (33:1) |
| 2 | Pd(TFA)2 instead of Pd(OAc)2 | 35 (33:1) |
| 3 | N-Ac-L-Leu-OH instead of N-Ac-Gly-OH | 40 (33:1) |
| 4 | Fmoc-Gly-OH instead of N-Ac-Gly-OH | 47(25:1) |
| 5 | Ag2CO3 instead of AgOAc | 72 (33:1) |
| 6 | AgTFA instead of AgOAc | 51 (33:1) |
| 7 | (CH2Cl)2 instead of HFIP | 33 (1:2) |
| 8 | TFE instead of HFIP | Trace |
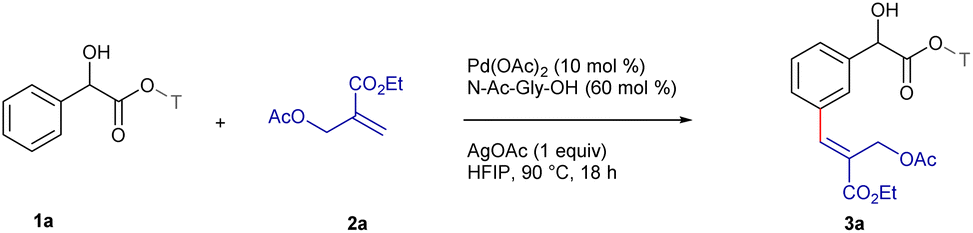
Having optimized the reaction conditions, we next explored the substrate scope using a series of α-substituted arylacetic acids utilizing 2a as the standard substrate (Scheme 1). Notably, the substrates bearing ortho-substituents such as chloro 1b and fluoro 1c reacted to deliver the meta-alkenylated products 3b and 3c in 72% and 68% yields, respectively, with ≥33:1 E/Z-selectivity. The reaction of meta-chloro substituted 1d gave 3d in 66% yield with an E/Z ratio of 16:1, while the substrates having chloro 1e and methoxy 1f groups at the para-position reacted to furnish 3e and 3f in 75% and 79% yields, respectively, with E-selectivity. Furthermore, 4-trifluoromethyl-substituted 1g coupled to furnish 3g in moderate yield with a 9:1 E/Z ratio, which might be due to the electron withdrawing nature of the substituent. However, the substrates with methyl 1j, ethyl 1k and methoxy 1l substituents at the α-position delivered the target scaffolds 3h–j in 66–80% yields with ≥8:1 E/Z-selectivity. In addition, bulky α-substituents, dimethyl 1m, diphenyl 1n, spirocyclohexyl 1o, spirocyclopentyl 1p, spirocyclopropyl 1q, and α-methyl-4-methyl 1r were compatible, furnishing 3k–p in 66–82% yields with ≥6:1 E/Z-selectivity. In contrast, the reaction of α-methyl-4-nitrophenylacetic acid 1s failed to deliver 3q, which might be due to the chelation of the nitro group with the Pd-catalyst. However, arylacetic acid 1t underwent reaction to afford 3r in 77% yield, albeit, with an E/Z ratio of 1.5:1. This may be attributed to the absence of α-substitution on the arylacetic acid, thereby leading to greater DG-meta distance. Furthermore, the reaction of substrates bearing ibuprofen 1u and ketoprofen 1v furnished 3s and 3t in 66% and 73% yields, respectively, with E-selectivity, while the reaction of glycopyrrolate and oxybutynin afforded 3u and 3v in 68% and 72% yields, respectively, with ≥20:1 E/Z-selectivity.
 | ||
| Scheme 1 Scope of α-substituted arylacetic acids.a,b,c,d aReaction conditions: 1 (0.1 mmol), 2a (0.12 mmol), Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (60 mol%), AgOAc (0.1 mmol), HFIP (1 mL), 90 °C, 18 h, air, pressure tube. bIsolated yield. cDetermined by 400 MHz 1H NMR. dn. d. = not detected. | ||
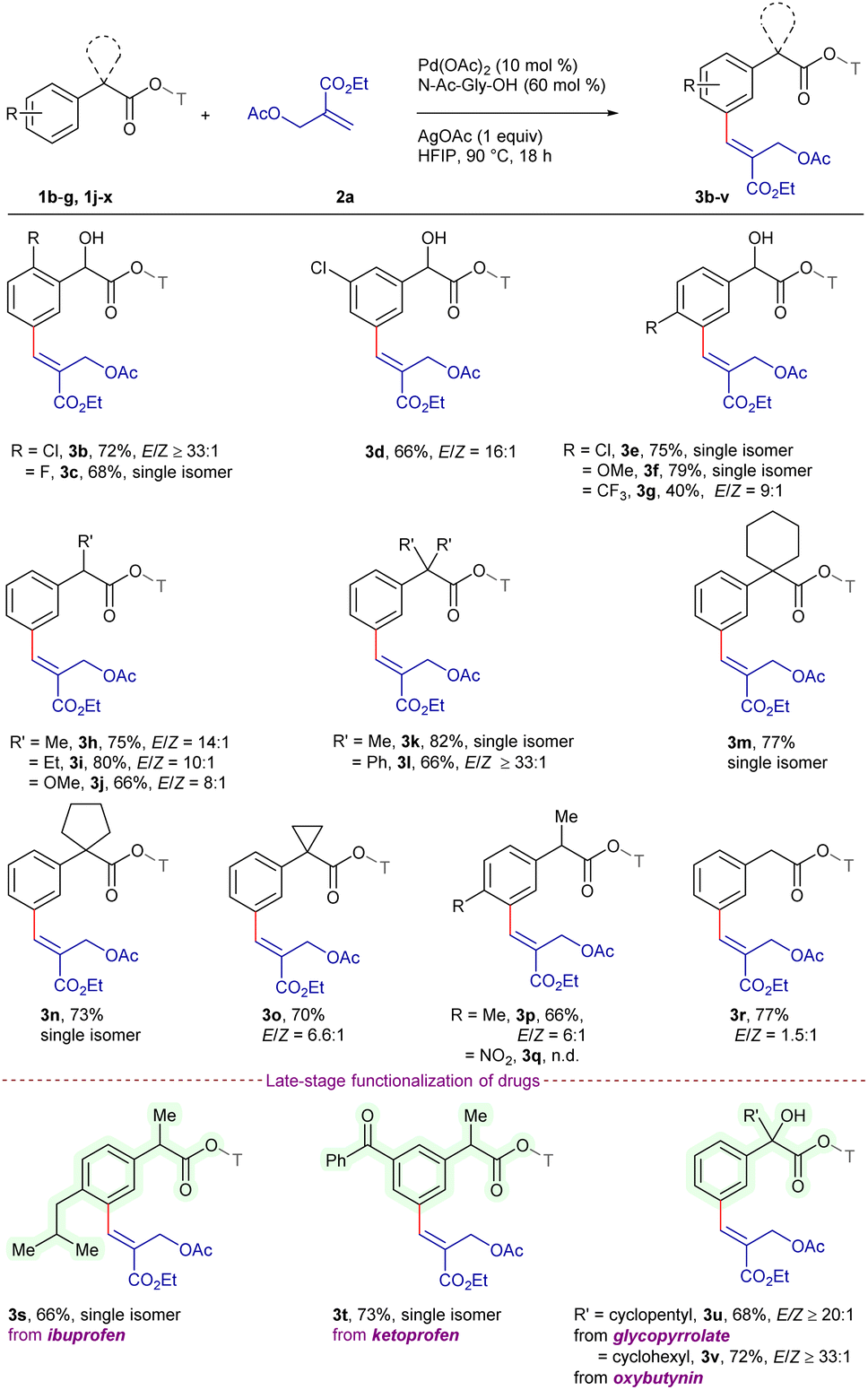
The scope of the procedure was extended for a series of MBH acetates 2b–h using 1m as the standard substrate (Scheme 2). MBH acetates bearing alkyl groups such as methyl 2b, benzyl 2c, cyclohexyl 2d and tert-butyl 2e demonstrated excellent compatibility, furnishing 3w–z in 69–80% yields with ≥20:1 E/Z-selectivity. Furthermore, Boc-protected 2f reacted to deliver 3aa in 63% yield, where the protecting group underwent hydrolysis to yield the alcohol. In addition, the reaction of MBH acetates derived from naturally occurring L-menthol 2g and (−)-borneol 2h gave 3ab and 3ac in 73% and 68% yields with ≥16:1 E/Z-selectivity, showcasing the synthetic potential for diversification of natural product derivatives.
 | ||
| Scheme 2 Scope of MBH acetates.a,b,c aReaction conditions: 1m (0.1 mmol), 2 (0.12 mmol), Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (60 mol%), AgOAc (0.1 mmol), HFIP (1 mL), 90 °C, 18 h, air, pressure tube. bIsolated yield. cDetermined by 400 MHz 1H NMR. | ||
Having established the meta-selective C–H functionalization, we turned our attention towards macrocyclic lactamization incorporating the arylacetic acid motif (Scheme 3). Gratifyingly, the macrocyclization proceeded employing Pd(OAc)2 (10 mol%), AgSbF6 (20 mol%) and H2O (1 equiv.) in (CH2Cl)2 at 100 °C for 8 h to deliver 4a in 71% yield with E-stereochemistry (Table S6, SI). A similar result was observed with α-disubstituted 3l to afford 4b in 78% yield with E-selectivity. This strategy could be extended to the substrates with benzyl 3x and cyclohexyl 3y substituents to produce 4c and 4d in 79% and 68% yields, respectively, with ≥20:1 E/Z-selectivity. Moreover, α-substituted arylacetic acids derived from ibuprofen 3s and ketoprofen 3t analogues reacted to produce 4e and 4f in 70% and 65% yields, respectively, with ≥13:1 E/Z-selectivity, whereas attempts to introduce structural complexity by replacing the α-methyl group with a hydroxy group resulted in a trace amount of 4g, which might be due to an unfavorable electronic effect impeding the cyclization. Furthermore, α-spirocyclopentyl 3n and α-spirocyclopropyl 3o underwent cyclization to afford 4h and 4i in 72 and 74% yields, respectively, with ≥14:1 E/Z-selectivity, highlighting the compatibility with sterically hindered spirocyclic substrates. Moreover, substrate 3a3 cyclized to deliver 4j in 76% yield with ≥20:1 E/Z selectivity, whereas 3a4 failed to furnish 4k, possibly due to less efficient formation of the key π-allyl palladium intermediate.
 | ||
| Scheme 3 Scope of macrocyclic lactams.a,b,c,d,e aReaction conditions: 3 (0.1 mmol), Pd(OAc)2 (10 mol%), AgSbF6 (20 mol%), H2O (0.1 mmol), (CH2Cl)2 (1 mL), 100 °C, 8 h, air, pressure tube. bIsolated yield. c3a3 has been used (please see Table S1). d3a4 has been used (please see Table S1). eDetermined by 400 MHz 1H NMR. | ||
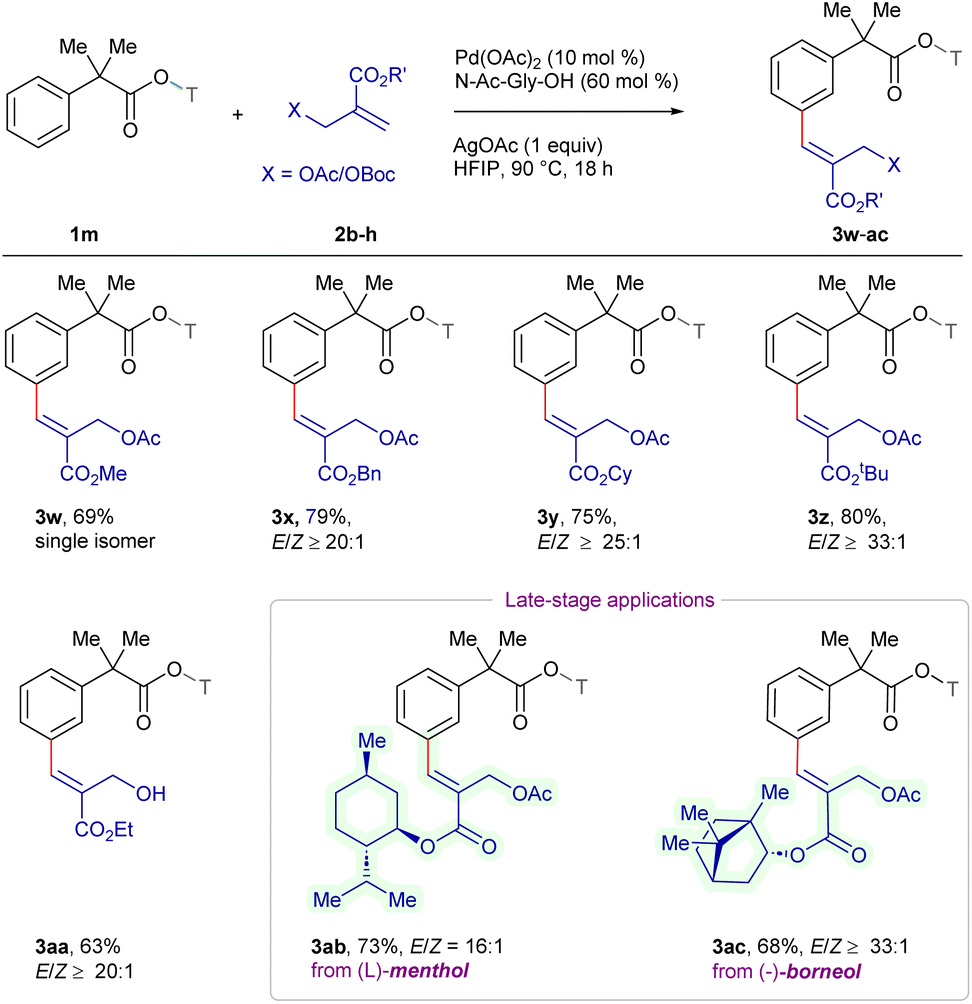
The scope of the procedure was further examined for the sequential meta/meta′ C–H difunctionalization employing 3x as a representative example to expand the synthetic utility (Scheme 4). Delightfully, 2,5-dihydrothiophene-1,1-dioxide 2i reacted to deliver 5 in 77% yield with ≥33:1 E/Z-selectivity. In addition, alkylation employing but-3-en-2-ol 2j furnished β-aryl ketone 6 in 72% yield with a 1:1 mixture of E/Z-isomers, which might be due to the non-conjugated nature of the alkyl group, while the reaction with 2a produced 7 in 80% yield with ≥33:1 E/Z-selectivity, demonstrating the versatility of procedure across multiple class of electrophiles.
 | ||
| Scheme 4 Scope of sequential distal C–H functionalization. aReaction conditions: 3x (0.1 mmol), 2i or 2a (0.12 mmol), Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (20 mol%), AgOAc (0.1 mmol), HFIP (1 mL), 90 °C, 18 h, air, pressure tube. b3x (0.1 mmol), 2j (0.12 mmol), Pd(OAc)2 (10 mol%), N-Ac-Gly-OH (20 mol%), Ag2CO3 (0.25 mmol), HFIP (1 mL), 90 °C, 18 h, air, pressure tube. cIsolated yield. | ||
To assess the scalability of the procedure, the reaction of 1m with 2c was investigated as a representative example on 1 mmol scale (Scheme 5). The reaction occurred to deliver 3x in 76% yield (304.8 mg). Furthermore, to demonstrate the removability of the nitrile template, hydrolysis of 3w was carried out utilizing LiOH in a mixture of THF, MeOH and H2O to provide carboxylic acid 8 in 78% yield, which can be hydrogenated to furnish meta-alkylated 9 in 80% yield. Moreover, the cross-coupling of 3x with phenyl boronic acid replaced the acetate moiety to afford 10 in 73% yield with an E/Z-selectivity of 5.2:1, while the acetate group was substituted with HFIP using NaOTf to furnish ether-linked 11 in 86% yield. In addition, the Pd-catalyzed azidation of 3k with NaN3 afforded 12 in 86% yield, which could be reduced via a Staudinger reaction to deliver amine derivative 13 in 69% yield, while a Cu(I)-catalyzed click reaction with phenylacetylene afforded triazole scaffold 14 in 89% yield.
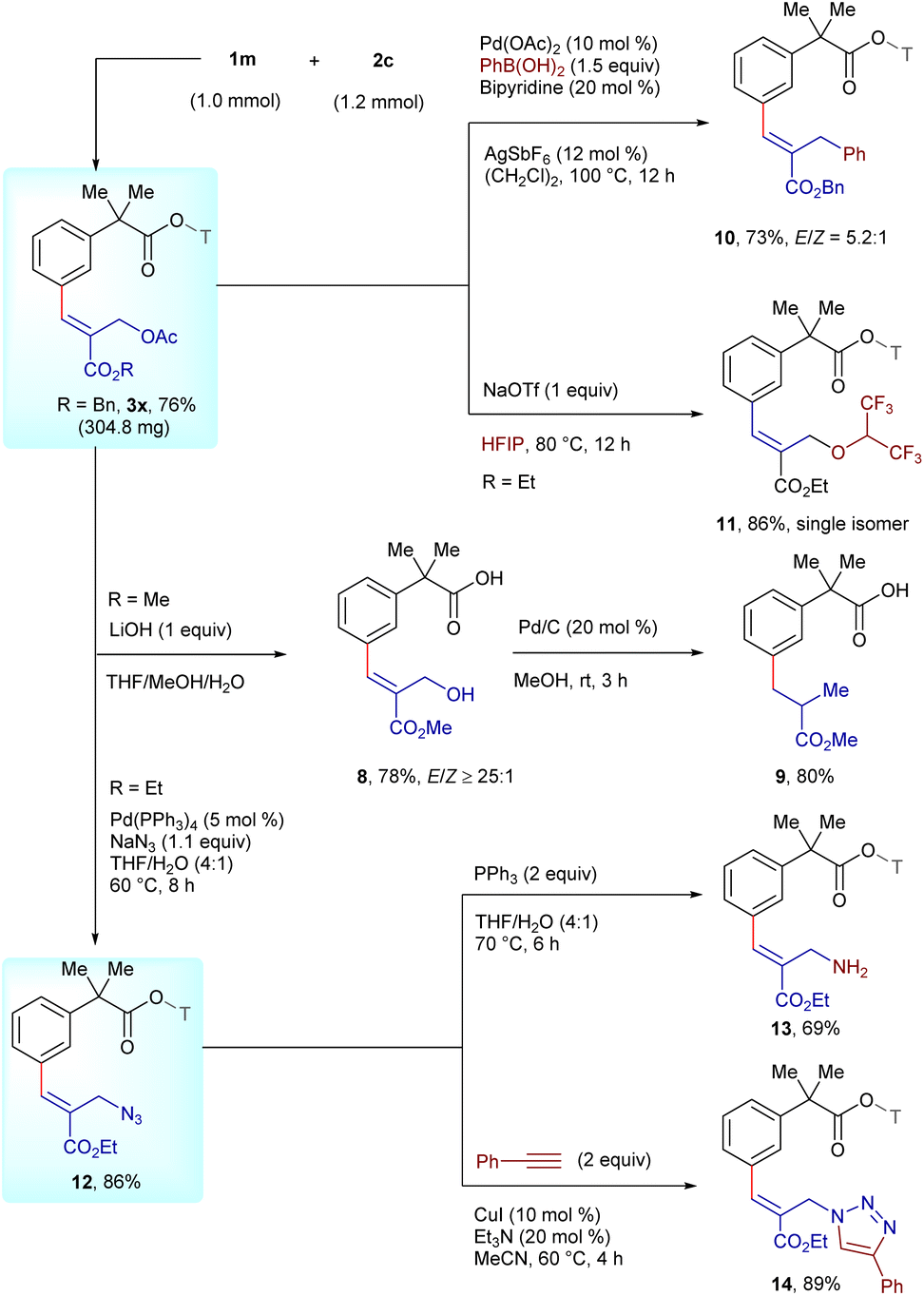 | ||
| Scheme 5 Removal of the template and synthetic utilities. | ||
To gain insight into the reaction pathway, deuterium labeling and kinetic isotope experiments were performed (Scheme 6). Treatment of substrate 1a with CD3CO2D resulted in 15% deuterium incorporation at the meta-position, indicating that C–H bond cleavage might be reversible (Scheme 6a). Furthermore, the intermolecular kinetic isotope experiment (KIE) of 1a with [D5]-1a exhibited kH/kD = 2.57, suggesting that C–H bond cleavage might be the rate-limiting step (Scheme 6b). Moreover, the reaction of arylacetic acid 1j′ with 2a employing 10 mol% Pd(OAc)2 and 1 equiv. Cu(OAc)2·H2O in (CH2Cl)2, followed by in situ esterification furnished 15 in 77% yield (Scheme 6c).15 This outcome suggests that the nitrile template plays a crucial role in selective C–H functionalization. In addition, the H218O experiment (detected via HRMS) suggests that water acts as the oxygen source in the macrocyclic lactam formation (Scheme 6d).
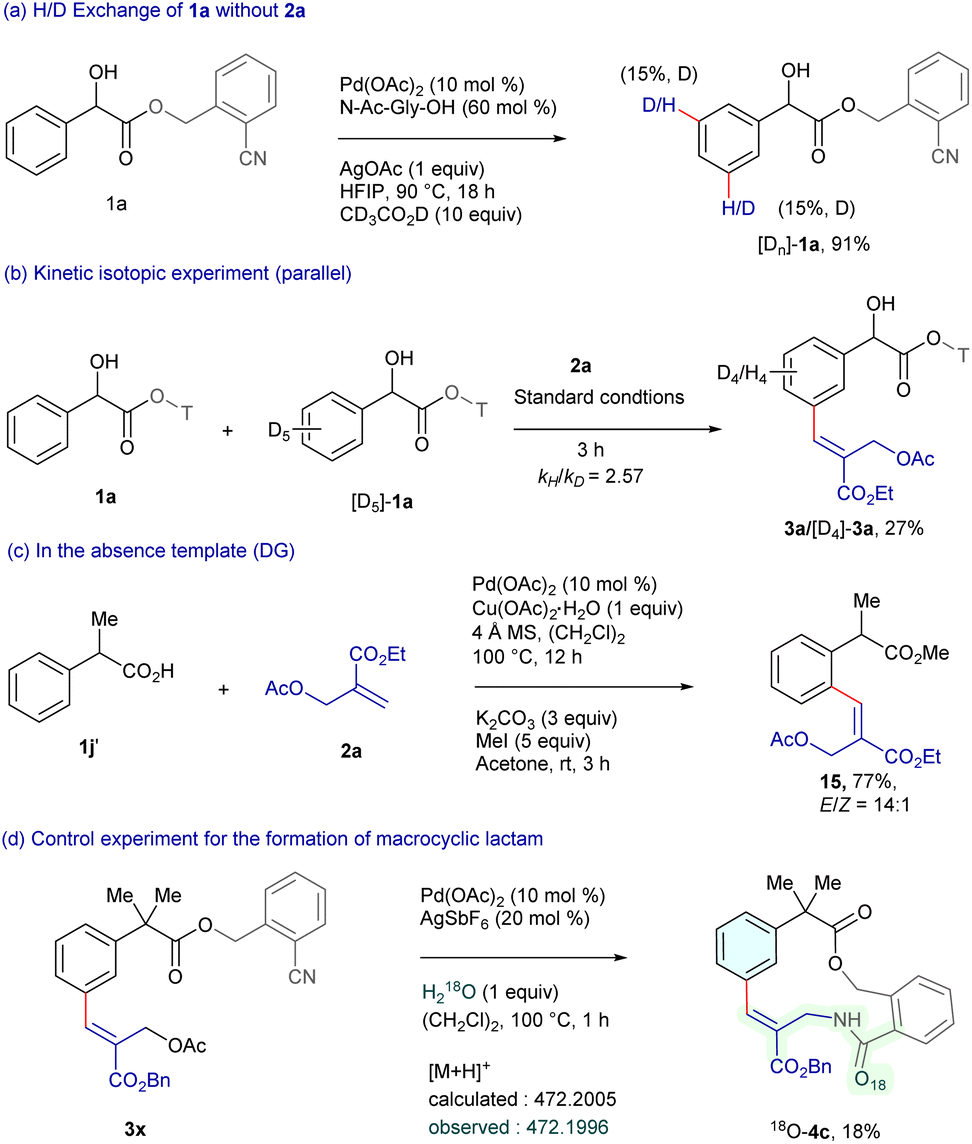 | ||
| Scheme 6 Mechanistic studies. | ||
The observed experimental results and literature precedent16 suggest that the substrate 1 can undergo a weak chelation with the Pd(II)-complex A to form B, bringing the metal in close proximity to the meta-C–H bond (cycle A, Scheme 7). Subsequently, the cleavage of the C–H bond can lead to the formation of palladacycle Cvia a concerted metalation and deprotonation (CMD) process. The coordination of C with the double bond of 2 can produce D that can lead a 1,2-migratory insertion to furnish E. The β-hydride elimination can yield the alkenylated 3 and Pd(II)-species, which can undergo reductive elimination to generate the Pd(0) species F and HX as a by-product. However, β-acetate elimination was not observed, likely due to the geometric and electronic constraints of metallacycle E (ref. 16c, 16d, 16f and 16g). The external oxidant AgOAc plays a crucial role in oxidizing Pd(0) to Pd(II), thereby facilitating the regeneration of the active Pd(II)-catalyst for the subsequent catalytic cycle.
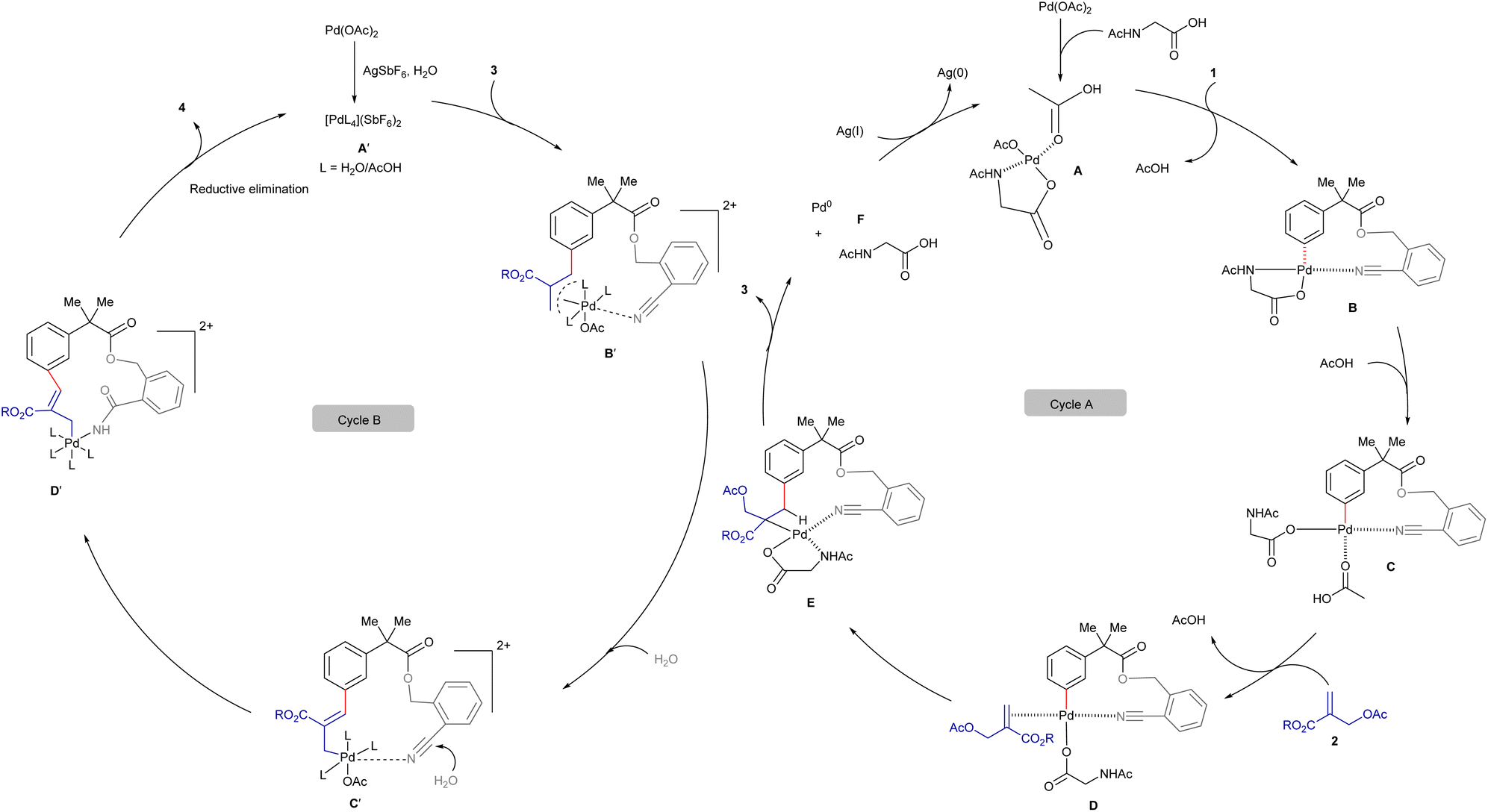 | ||
| Scheme 7 Proposed catalytic cycle. | ||
The second catalytic cycle of the macrocyclization is depicted in cycle B (Scheme 7).17 Initially, the Pd(II)-species A′ reacts with 3 to produce the Pd-π-allyl-complex B′ that can lead to the formation of the palladacycle C′. Hydration of the chelated C–N triple bond can produce D′ that can undergo reductive elimination to deliver the 14-membered lactam 4, concurrently regenerating the Pd(II)-species A′.
The photophysical properties of the selected macrocyclic lactams were investigated due to their conjugated framework. UV-vis and fluorescence studies (in CH3CN) revealed emission maxima at λem 340 nm (Fig. S2, SI), indicating the high-energy excited state characteristic of the aromatic macrocycles. Notably, lactam 4a exhibited a red-shifted emission, which suggests the intramolecular charge transfer (ICT), while the products 4c–f showed negligible shifts, implying the limited ICT contributions.18
Furthermore, the biocompatibility and cytotoxicity of 4a and 4e–f were evaluated as representative examples using MTT assays against Vero and MCF-7 cells. Vero cells showed no significant morphological changes or viability loss up to 200 µg mL−1, revealing their non-toxic nature, while the cytotoxicity assays on MCF-7 cells showed dose-dependent morphological changes and reduced viability at higher concentrations. Overall, the lactams were biocompatible with normal cells yet cytotoxic to cancer cells, highlighting their therapeutic potential (Fig. S3–5 and Table S7, SI).
Conclusion
In summary, we have developed a Pd-catalyzed directed meta-selective C–H functionalization of α-substituted arylacetic acids with MBH acetates. The β-aryl MBH acetate can subsequently be cyclized to produce 14-membered macrocyclic lactams. The procedure can be extended to achieve difunctionalization with varied coupling partners. The substrate scope, meta-selectivity, functional group diversity, photophysical properties, biological activities and post-synthetic applications are the important practical features.Author contributions
P. M.: designed, performed, analysed the experiments and writing the manuscript. P. K. M., V. T. and S. K.: analysed the experiments and writing the manuscript. T. P.: conceptualization, funding acquisition, investigation, project administration, supervision, visualization, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, additional figures, tables, photophysical study, cytotoxicity study and spectral data. See DOI: https://doi.org/10.1039/d5sc08286b.Acknowledgements
We thank SERB (SCP/2022/000256 and CRG/2022/002778) for financial support, CIF, Chemistry (SR/FST/CSII/2017/23C) and NECBH (BT/NER/143/SP44675/2023) for NMR, HRMS and XRD facilities. P. M. (PDF/2023/000362), P. K. M. (INSPIRE), V. T. (IPDF), S. K. (PMRF) and T. P. (JCB/2022/000037) thank SERB, DST, IIT Guwahati and MoE for their fellowships.Notes and references
- For reviews, see: (a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (b) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (c) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481 CrossRef CAS PubMed; (d) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS; (e) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (f) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304 CrossRef CAS PubMed; (g) G. Liao, T. Zhang, Z. K. Lin and B. F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773 CrossRef CAS; (h) N. Y. S. Lam, K. Wu and J. Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767 CrossRef CAS; (i) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed; (j) B. Zhao, B. Prabagar and Z. Shi, Chem, 2021, 7, 2585 CrossRef CAS.
- For reviews, see: (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (b) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798 CrossRef CAS; (c) B. Li, A. I. M. Ali and H. Ge, Chem, 2020, 6, 2591 CrossRef CAS; (d) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399 CrossRef CAS; (e) C. M. Josephitis, H. M. H. Nguyen and A. McNally, Chem. Rev., 2023, 123, 7655 CrossRef CAS PubMed; (f) P. Bellotti, H. M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237 CrossRef CAS PubMed; (g) I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619 CrossRef CAS PubMed; (h) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692 CrossRef CAS; (i) Y. F. Liang, M. Bilal, L. Y. Tang, T. Z. Wang, Y. Q. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Chem. Rev., 2023, 123, 12313 CrossRef CAS PubMed.
- For reviews, see: (a) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed; (b) J. H. Mao, Y. Bin Wang, L. Yang, S. H. Xiang, Q. H. Wu, Y. Cui, Q. Lu, J. Lv, S. Li and B. Tan, Nat. Chem., 2021, 13, 982 CrossRef CAS; (c) B. Prabagar, Y. Yang and Z. Shi, Chem. Soc. Rev., 2021, 50, 11249 RSC; (d) B. Li, M. Elsaid and H. Ge, Chem, 2022, 8, 1254 CrossRef CAS; (e) K. Yamatsugu and M. Kanai, Chem. Rev., 2023, 123, 6793 CrossRef CAS PubMed; (f) J. Hu, S. Pradhan, S. Waiba and S. Das, Chem. Sci., 2024, 16, 1041 RSC; (g) G. Meng, J. L. Yan, N. Chekshin, D. A. Strassfeld and J. Q. Yu, ACS Catal., 2024, 14, 12806 CrossRef CAS PubMed.
- For reviews, see: (a) J. Zhang, X. Lu, C. Shen, L. Xu, L. Ding and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263 RSC; (b) Q. Zhang and B. F. Shi, Chem. Sci., 2021, 12, 841 RSC.
- For reviews, see: (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS; (b) S. Rej, A. Das and N. Chatani, Coord. Chem. Rev., 2021, 431, 213683 CrossRef CAS; (c) C. Haldar, M. E. Hoque, J. Chaturvedi, M. M. M. Hassan and B. Chattopadhyay, Chem. Commun., 2021, 57, 13059 RSC; (d) R. L. Pilkington and D. L. Priebbenow, ACS Catal., 2025, 15, 6881 CrossRef CAS.
- (a) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J. Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571 CrossRef CAS; (b) N. Y. S. Lam, Z. Fan, K. Wu, H. S. Park, S. Y. Shim, D. A. Strassfeld and J. Q. Yu, J. Am. Chem. Soc., 2022, 144, 2793 CrossRef CAS.
- (a) X. C. Wang, W. Gong, L. Z. Fang, R. Y. Zhu, S. Li, K. M. Engle and J. Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed; (b) L. Y. Liu, J. X. Qiao, K. S. Yeung, W. R. Ewing and J. Q. Yu, J. Am. Chem. Soc., 2019, 141, 14870 CrossRef CAS PubMed; (c) J. Chaturvedi, C. Haldar, R. Bisht, G. Pandey and B. Chattopadhyay, J. Am. Chem. Soc., 2021, 143, 7604 CrossRef CAS PubMed; (d) W. Chang, Y. Chen, S. Lu, H. Jiao, Y. Wang, T. Zheng, Z. Shi, Y. Han, Y. Lu, Y. Wang, P. Pan, J. Q. Yu, K. N. Houk, F. Liu and Y. Liang, Chem, 2022, 8, 1775 CrossRef CAS; (e) A. Mondal, M. Díaz-Ruiz, F. Deufel, F. Maseras and M. van Gemmeren, Chem, 2023, 9, 1004 CrossRef CAS PubMed; (f) M. Schnürch, Chem, 2023, 9, 764 CrossRef; (g) V. Sukowski, M. van Borselen, S. Mathew, B. de Bruin and M. Á. Fernández-Ibáñez, Angew. Chem., Int. Ed., 2024, 63, e202317741 CrossRef CAS PubMed.
- (a) L. Wan, N. Dastbaravardeh, G. Li and J. Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056 CrossRef CAS; (b) L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J. Q. Yu, ACS Cent. Sci., 2015, 1, 394 CrossRef CAS PubMed; (c) Y. F. Yang, X. Hong, J. Q. Yu and K. N. Houk, Acc. Chem. Res., 2017, 50, 2853 CrossRef CAS PubMed.
- (a) R. Y. Tang, G. Li and J. Q. Yu, Nature, 2014, 507, 215 CrossRef CAS; (b) C. G. Frost and A. J. Paterson, ACS Cent. Sci., 2015, 1, 418 CrossRef CAS PubMed; (c) H. J. Xu, Y. Lu, M. E. Farmer, H. W. Wang, D. Zhao, Y. S. Kang, W. Y. Sun and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200 CrossRef CAS PubMed; (d) H. J. Xu, Y. S. Kang, H. Shi, P. Zhang, Y. K. Chen, B. Zhang, Z. Q. Liu, J. Zhao, W. Y. Sun, J. Q. Yu and Y. Lu, J. Am. Chem. Soc., 2019, 141, 76 CrossRef CAS PubMed; (e) S. Li, H. Wang, Y. Weng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 18502 CrossRef CAS; (f) B. Wang, Y. Zhou, N. Xu, X. Xu, X. Xu and Z. Jin, Org. Lett., 2019, 21, 1885 CrossRef CAS PubMed; (g) D. Srinivas and G. Satyanarayana, Org. Lett., 2021, 23, 7353 CrossRef CAS PubMed; (h) P. Muthuraja, R. Usman, R. Sajeev and P. Gopinath, Org. Lett., 2021, 23, 6014 CrossRef CAS PubMed; (i) H. Wang, H. Li, X. Chen, C. Zhou, S. Li, Y. F. Yang and G. Li, ACS Catal., 2022, 12, 13435 CrossRef CAS; (j) P. Gupta, S. Madhavan and M. Kapur, Angew. Chem., Int. Ed., 2023, 62, e202305278 CrossRef CAS; (k) M. Bakthadoss and T. T. Reddy, Chem. Sci., 2023, 14, 5880 RSC; (l) Y. L. Zhu, Q. X. Sun, J. S. Feng, Y. D. Shao and J. Chen, Org. Lett., 2023, 25, 4416 CrossRef CAS PubMed; (m) M. Jeganmohan and A. Dutta, Chem.–Eur. J., 2024, 30, e202402162 CrossRef; (n) K. Mounika and G. Satyanarayana, Org. Lett., 2024, 26, 8899 CrossRef CAS PubMed; (o) C. Sreenivasulu, P. Ramesh, D. R. Kishore, D. Srinivas and G. Satyanarayana, Org. Lett., 2025, 27, 6599 CrossRef CAS PubMed.
- P. Zhang, Z. Jiang, Z. Fan, G. Li, Q. Ma, J. Huang, J. Tang, X. Xu, J. Q. Yu and Z. Jin, Chem. Sci., 2023, 14, 8279 RSC.
- (a) L. H. Tu, H. Noor, P. Cao and D. P. Raleigh, ACS Chem. Biol., 2014, 9, 1632 CrossRef CAS; (b) T. Lynagh, J. L. Romero-Rojo, C. Lund and S. A. Pless, J. Med. Chem., 2017, 60, 8192 CrossRef CAS PubMed; (c) F. Noverraz, E. Montanari, J. Pimenta, L. Szabó, D. Ortiz, C. Gonelle-Gispert, L. H. Bühler and S. Gerber-Lemaire, Bioconjug. Chem., 2018, 29, 1932 CrossRef CAS PubMed.
- (a) G. Liao, B. Li, H. M. Chen, Q. J. Yao, Y. N. Xia, J. Luo and B. F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151 CrossRef CAS PubMed; (b) A. K. Pandey, S. H. Han, N. K. Mishra, D. Kang, S. H. Lee, R. Chun, S. Hong, J. S. Park and I. S. Kim, ACS Catal., 2018, 8, 742 CrossRef CAS; (c) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476 CrossRef CAS; (d) S. Pradhan, P. B. De and T. Punniyamurthy, Org. Lett., 2019, 21, 9898 CrossRef CAS PubMed; (e) H. Wang, Z. Li, X. Chen, J. J. Wong, T. Bi, X. Tong, Z. Xu, M. Zhen, Y. Wan, L. Tang, B. Liu, X. Zong, D. Xu, J. Zuo, L. Yang, W. Huang, K. N. Houk and W. Yang, Chem, 2023, 9, 607 CrossRef CAS.
- Macrocyclization using cycloaddition, ring-closing metathesis and condensation, see: (a) E. A. Crane and K. A. Scheidt, Angew. Chem., Int. Ed., 2010, 49, 8316 CrossRef CAS PubMed; (b) X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540 CrossRef CAS PubMed; (c) K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed; (d) K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288 CrossRef CAS PubMed; (e) S. Huang, Z. Lei, Y. Jin and W. Zhang, Chem. Sci., 2021, 12, 9591 RSC; (f) H. Zhai, K. Lv, J. Li, J. Wang, T. Liu and C. Zhao, J. Am. Chem. Soc., 2024, 146, 29214 CrossRef CAS PubMed.
- (a) B. Over, S. Wetzel, C. Grütter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Nat. Chem., 2013, 5, 21 CrossRef CAS PubMed; (b) L. R. Malins, J. N. Degruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233 CrossRef CAS PubMed; (c) A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167 CrossRef CAS PubMed; (d) S. Ren, G. Y. Qiao and J. R. Wu, Chem. Soc. Rev., 2024, 53, 10312 RSC; (e) W. Xu and C. Kang, J. Med. Chem., 2025, 68, 5000 CrossRef CAS PubMed.
- M. Z. Lu, X. R. Chen, H. Xu, H. X. Dai and J. Q. Yu, Chem. Sci., 2018, 9, 1311 RSC.
- (a) H. Ohmiya, Y. Makida, D. Li, M. Tanabe and M. Sawamura, J. Am. Chem. Soc., 2010, 132, 879 CrossRef CAS PubMed; (b) Y. F. Yang, G. J. Cheng, P. Liu, D. Leow, T. Y. Sun, P. Chen, X. Zhang, J. Q. Yu, Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed; (c) J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746 RSC; (d) B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, A. C, S. Malakar, H. F. Schaefer and R. B. Sunoj, Chem. Sci., 2020, 11, 208 RSC; (e) G. Li, Y. Yan, P. Zhang, X. Xu and Z. Jin, ACS Catal., 2021, 11, 10460 CrossRef CAS; (f) M.-H. Yang and R. A. Altman, Nat. Synth., 2022, 1, 753 CrossRef CAS; (g) M. K. Bogdos, O. Stepanović, A. Bismuto, M. G. Luraschi and B. Morandi, Nat. Synth., 2022, 1, 787 CrossRef CAS.
- T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949 CrossRef CAS.
- P. S. K. Prabhakar Ganesh, P. Muthuraja and P. Gopinath, Chem. Commun., 2022, 58, 4211 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |