
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStapled histone H3 tails are super-substrates for lysine methyltransferase SETD7
Nurgül
Bilgin
 ,
Laust
Moesgaard
,
Jacob
Kongsted
and
Jasmin
Mecinović
*
,
Laust
Moesgaard
,
Jacob
Kongsted
and
Jasmin
Mecinović
*
Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense, Denmark. E-mail: mecinovic@sdu.dk
First published on 2nd December 2025
Abstract
The SETD7-catalysed methylation of lysine 4 in histone 3 (H3K4) plays an important role in the epigenetic control of eukaryotic gene expression. The N-terminal tail of histone H3 binds to SETD7 in a bend-like conformation in which Ala1 and Thr6 are located in close proximity, enabling the Lys4 substrate residue to react with the methyl group of the S-adenosylmethionine cosubstrate. Here, we report a proximity-guided design of H3 peptides stapled between amino acid residues 1 and 6 as potential substrates and inhibitors of human SETD7. Our results demonstrate that most of the appropriately stapled H3 peptides are efficiently methylated by SETD7, outperforming the unstructured, linear histone H3 tail sequence found in nature. The cyclic H3 peptides possessing the lactam linkage are excellent SETD7 substrates, outcompeting the linear H3K4 peptide, as demonstrated by up to 110-fold increase in catalytic efficiencies. The stapled H3 peptides display exclusive substrate selectivity for SETD7 over related H3K4 methyltransferases MLL3 and SETD1A. Inhibition assays show that the norleucine variant of the most efficient 1,6-stapled peptide substrate is a potent inhibitor of human SETD7. Overall, the results highlight a novel approach to selectively modulate the SETD7 activity and emphasise the potential of stapled histone peptides as exceptionally efficient peptidomimetic substrates and inhibitors of epigenetic enzymes.
Introduction
In eukaryotes, DNA is packed into chromatin. Core histone proteins (H2A, H2B, H3 and H4) assemble into an octameric complex around which about 147 base pairs of DNA wrap, forming the nucleosome, the central repeating unit of the eukaryotic chromatin.1–3 The unstructured N-terminal histone tails, which protrude from the nucleosome, are subject to various posttranslational modifications (PTMs) that regulate the structure and function of chromatin.1,4 Histone lysine methylations are key PTMs that play important roles in the epigenetic regulation of eukaryotic gene expression.4,5 Lysine methylation marks are extensively found at various sites on histone proteins, and their cellular levels are enzymatically regulated by histone lysine methyltransferases (KMTs) and histone lysine demethylases (KDMs).4,6–8 Methylated lysine residues on histones can exist in three different states: mono-, di- and trimethyllysine (Kme, Kme2, and Kme3), each maintaining the positive charge of lysine side chain.6,9 The histone methylation sites and states influence chromatin dynamics, and can result in both transcriptional activation and repression.10Su(var) Enhancer of Zeste and Trithorax (SET) domain enzymes are characterised as methyltransferases responsible for specific methylation of lysine residues in histone and non-histone proteins, and can be classified in accord to the lysine residue that they target, with their dysregulation linked to several human diseases.6,11,12 SET-domain containing protein 7 (SETD7, SET7/9, KMT7) is a histone lysine methyltransferase that catalyses the transfer of a single methyl group on the Nε-amino group of lysine 4 on histone 3 tail (H3K4) by using the S-adenosylmethionine (SAM) cosubstrate as the methyl donor, forming the H3K4me product and the S-adenosylhomocysteine (SAH) byproduct (Fig. 1A).13–15 H3K4 methylation is a histone mark that activates transcription, however, mutations in H3K4 methyltransferases can impact related cellular processes in health and disease.16,17 Other histone lysine methyltransferases include MLL1-4 and SETD1A/B, which can install different methyl marks on H3K4.6,18–20 Overexpression of SETD7 is associated with various cancer types21–23 and affects other cellular events.24–26 To this end, potent small molecule inhibitors targeting SETD7 have been recently developed.27–29
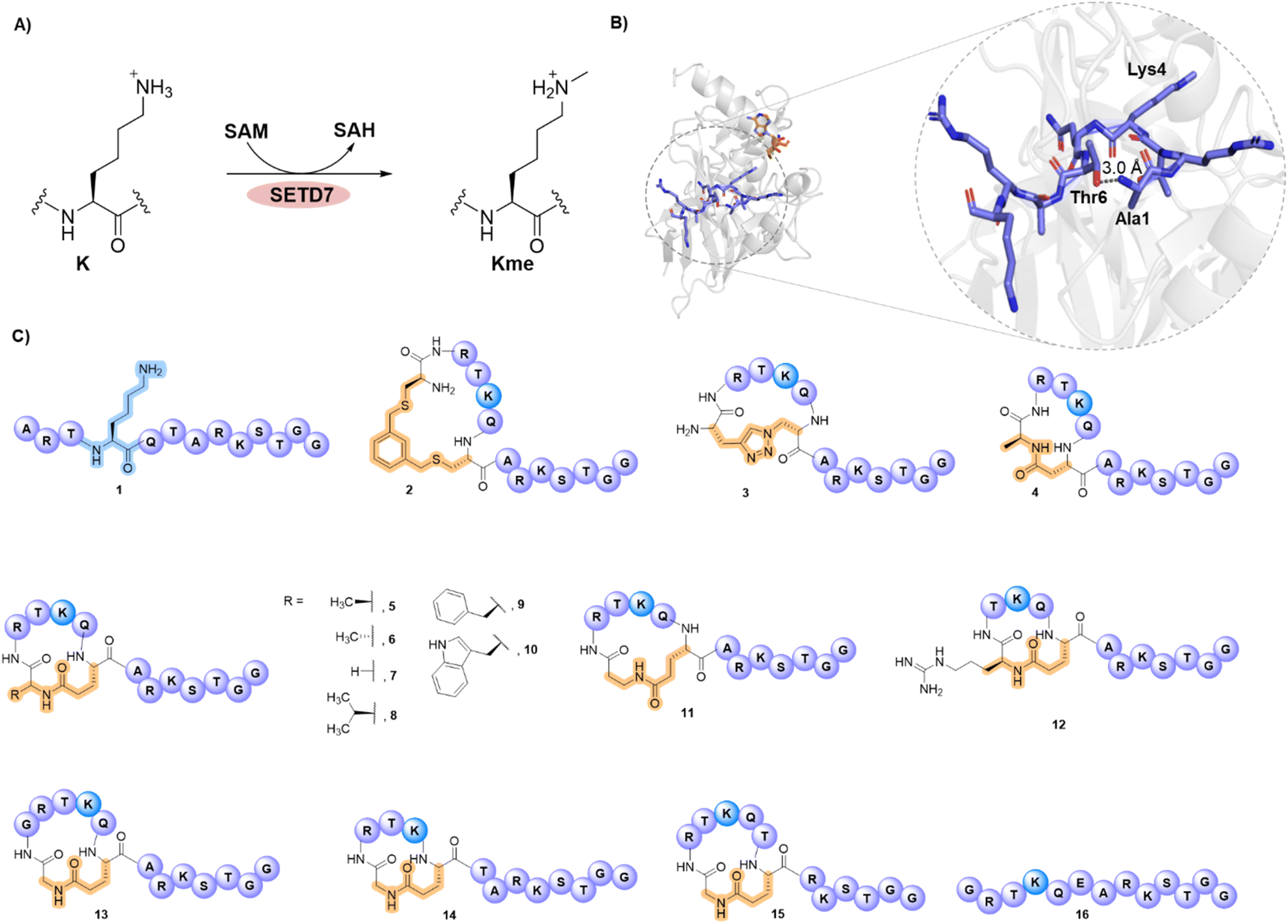 | ||
| Fig. 1 SETD7-catalysed methylation of H3K4. (A) Monomethylation of lysine 4 residue by methyltransferase SETD7 in the presence of the SAM cosubstrate. (B) View from a crystal structure of SETD7 (gray) in complex with the H3K4me peptide (blue) and SAH (orange) (PDB: 1O9S), and a zoomed view on the H3K4me peptide structure in a bend-like conformation, highlighting the main-chain-to-side-chain distance between the proximate Ala1 and Thr6 residues. (C) A panel of designed stapled H3 peptides as potential substrates for SETD7 (2–15); linear H3 peptides 1 and 16 are used as controls for comparison. | ||
The structure of the ternary complex SETD7-H3K4me-SAH revealed that residues belonging to the SET domain (residues 337–349) form a β-hairpin structure and accommodate the H3 peptide substrate that adopts a sharply bended conformation upon binding.15 Leu267 and Tyr residues (305, 335 and 337) of SETD7 form the peptide binding groove from which the lysine substrate protrudes (Fig. 1B), facilitating hydrophobic interactions with the alkyl chain of lysine and H-bonding interactions with the ε-amino group.15 The lysine ε-amino group acts as a H-bond donor to Tyr245 and Tyr335 at the active site, which take part in deprotonation and stabilise the orientation of lysine towards the methyl group of SAM for efficient methyl transfer reaction.15,30 Mutation of the Tyr245 residue in SETD7 was not only shown to reduce the methyltransferase activity, but also disrupts the arrangement of the protein structure, releasing a water molecule involved in the enzymatic methylation process.15
Stapling of peptides has emerged as a chemical strategy that can provide superior protein binders, often resulting in enhanced biological activity.31 Compared to their linear counterparts, stapled peptides have rigid and locked structures, leading to enhanced binding affinity, higher target specificity and improved biostability.32 These features conspire to make stapled peptides an important class of protein–protein interaction modulators. However, the ability to generate such peptides against biomedical targets without available biostructural information regarding protein interactions, mechanism of action or binding sites remains difficult. Current strategies rely on the RaPID platform that can typically generate large libraries of macrocyclic peptides and has been applied to various proteins.31,33–35 In this work we highlight the potential of a proximity-guided stapling strategy as a general concept that can be applied to design peptide substrates and modulators of epigenetic proteins.
The notable feature of the bent H3 peptide in a complex with SETD7 inspired us to design and develop a series of stapled H3 peptides via the covalent linkage of the two proximal residues (Ala1 and Thr6) (Fig. 1B and C). We hypothesised that stapling of residues 1 and 6 in histone H3 peptides might lead to histone substrates that outperform the linear H3 peptide possessing the natural sequence. While there is no example of a cyclic peptide outcompeting the naturally-occurring histone substrate of any epigenetic writer enzyme, engineered linear super-substrate peptides were recently developed, modulating the NSD2 and SETD2 methyltransferase activity.36–38 Herein, we apply synthetic methods to cyclise H3 peptides and showcase the discovery of stapled H3 peptides for exceptionally efficient SETD7 catalysis and inhibition.
Results and discussion
The structure of the SETD7-H3K4me-SAH complex suggests that the conformational constraint induced by the H3K4 stapling through the covalent linking of proximal positions Ala1 and Thr6 might preorganise/stabilise the histone H3 tail peptide structure and exhibit high affinity and selectivity for SETD7 (Fig. 1B). We selected Ala1 and Thr6 sites for stapling as they appear to be in close proximity in a “head-to-side-chain” arrangement: the OH group of Thr6 has a distance of 3.0 Å to the N-terminal amino group of Ala1, and the shortest distance of 2.8 Å to the side-chain methyl carbon of Ala1. The β-carbon of Thr6 has a distance of 3.8 Å to the side-chain methyl carbon and the N-terminal group of Ala1. We started the investigations by synthesising a panel of 13-mer H3 peptides (residues 1–13, ARTKQTARKSTGG) stapled at positions 1 and 6 employing three different stapling strategies: bis-thiol alkylation,39 azide–alkyne CuI-mediated Huisgen 1,3-dipolar cycloaddition,40 and lactam formation41 (Fig. 1C). Based on the proximity of the Ala1 and Thr6 residues leading to a bend-like conformation of the H3K4 peptide in the SETD7 bound state, we further conceived that the amide bond formation strategy allows the introduction of a broader stapled peptide scope via substitution of Ala1 by different canonical and non-canonical amino acids, providing deeper insight into the efficiency and selectivity of SETD7. The stapled peptides 2–15 were synthesised employing Fmoc-based solid-phase peptide synthesis (SPPS) and subsequent in-solution side-chain-to-side-chain or on-resin head-to-side-chain cyclisation. Purity and characterisation of all synthetic histone peptides were assessed by MALDI-TOF MS and analytical HPLC (Table S1 and Fig. S1–S24).Initial assays were carried out by the incubation of recombinant human SETD7 (500 nM) in the presence of a synthetic stapled or linear H3K4 peptide 1–16 (10 µM) and SAM (30 µM) in assay buffer (50 mM HEPES, 0.1 mM EDTA, 1 mM DTT, pH 8.0) at 37 °C. Potential conversion to methylated products (+14 Da mass shift) was monitored after 1 and 3 h by MALDI-TOF MS (Fig. 2, S25 and S26). Under these conditions, the level of methylation of the ‘native’ linear H3K4 substrate (1) by SETD7 was observed to be ∼56% after 1 h, reaching ∼82% methylation after 3 h (Fig. 2A and S25). The enzyme assays revealed that the stapled bis-thioether (2) and triazole (3) H3 peptides were methylated to a lesser extent (∼65% and ∼78%, respectively) by SETD7 after 3 h (Fig. 2A), which can be attributed to more rigid and/or bulkier linkages. Lactam-based H3A1-D6 (4) and H3A1-E6 (5) peptides, however, were nearly completely methylated within 3 h (Fig. 2A and S26). Our enzymatic assays also showed that H3A1-D6 (4) was methylated to a lower extent (∼63%), while H3A1-E6 (5) was fully converted to methylated product after 1 h (Fig. S25), indicating that Glu6 possesses a better side-chain length for further design of stapled peptides and that a shorter ring size is less favourable for productive binding and SETD7 catalysis. Notably, changing the stereochemistry of the cyclising N-terminal residue as for D-Ala (6) did not affect the catalytic activity of SETD7 (Fig. 2A). The stapled peptide variants of N-terminal Ala1 substituted by Gly (7), Val (8), Phe (9), Trp (10) and βAla (11), were also efficiently converted (≥90%) to the corresponding methylated products within 3 h (Fig. 2A and S26), indicating that non-bulky and bulky groups can be accommodated for efficient SETD7 binding and catalysis. A stapled shorter (lacking Ala1, 12) or extended (with addition of Gly0, 13) peptides were observed to yield ∼16% and ∼65% methylated product, respectively, after 3 h reaction (Fig. 2A and S26). The stapled H3G1-E5 (14) and H3G1-E7 (15) peptides did not undergo methylation by SETD7 within the detection limits, suggesting that stapling requires the connection between residues 1 and 6 for efficient SETD7 catalysis (Fig. 2A and S26). Notably, the linear control peptide H3G1E6 (16) was methylated at low levels (∼36%) by SETD7 after 3 h, indicating that the flexible conformation of the peptide structure is less favoured compared to the stapled counterpart (7) (Fig. 2A). Taken together, these results indicate that the X1-E6 amide bond stapled peptides are superior SETD7 substrates over the linear and other cyclic H3 peptides.
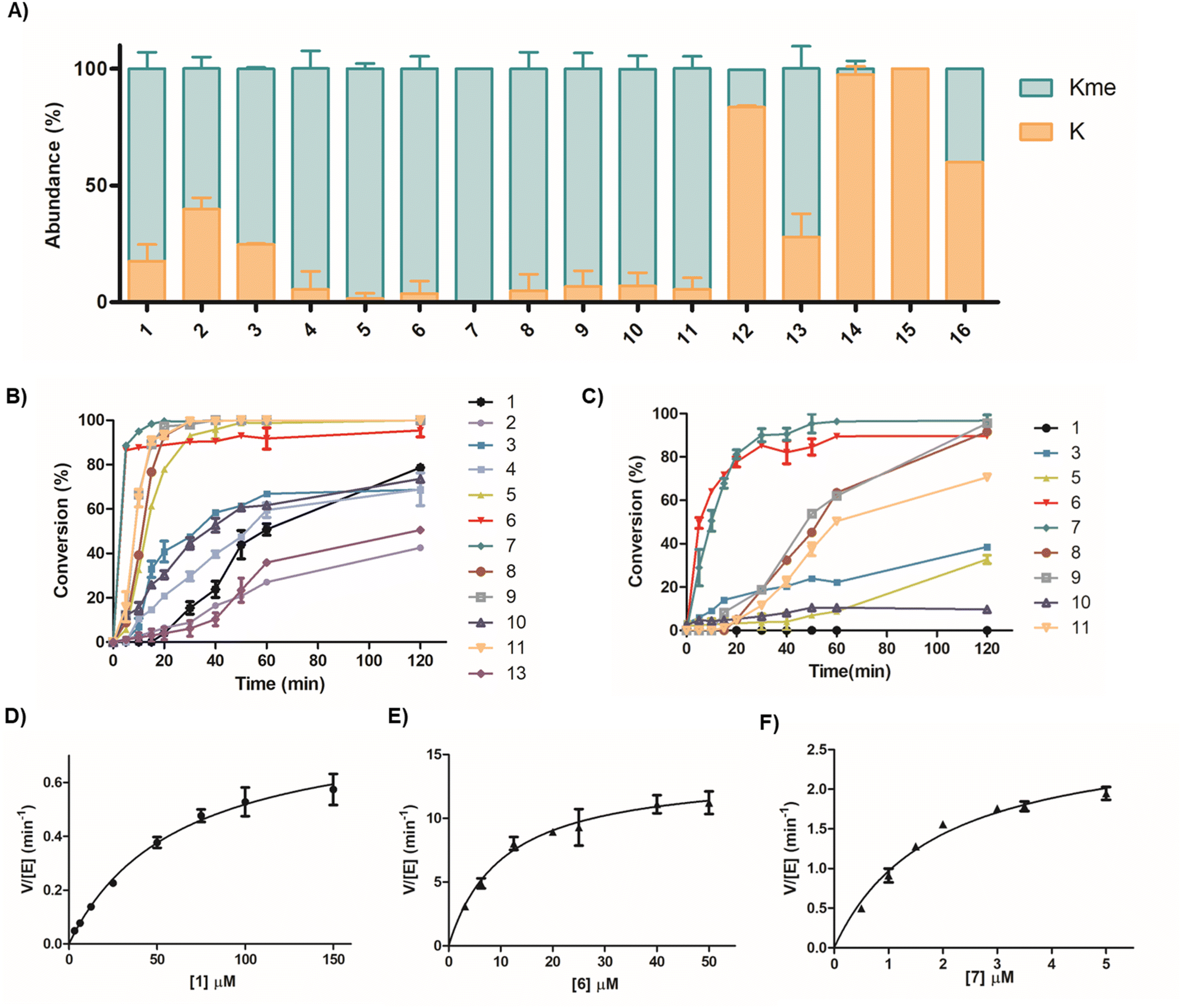 | ||
| Fig. 2 Time-course and steady-state kinetics of the SETD7-catalysed methylation of H3 peptides using MALDI-TOF MS assays. (A) Conversion of SETD7-catalysed (500 nM) methylation of linear H3K4 substrates and stapled H3 peptides (10 µM) in the presence of SAM (30 µM) after 3 h at 37 °C. (B) Time-course data of the SETD7-catalysed (500 nM) methylation of linear and stapled H3 peptides (10 µM). Error bars are reported as standard deviations from two independent replicates (n = 2). (C) Time-course data of the SETD7-catalysed (100 nM) methylation of linear and stapled H3 peptides (10 µM) in the presence of SAM (30 µM). Error bars are reported as standard deviations from two independent replicates (n = 2). (D) Kinetics plot for the linear H3K4 (1) substrate. (E) Kinetics plot for the stapled peptide 6. (F) Kinetics plot for the stapled peptide 7. Error bars for kinetics plots are reported as standard deviations from three independent replicates (n = 3). | ||
To further investigate the catalytic efficiency of SETD7 for the poorer H3 substrates, assays with a higher SETD7 concentration (1 µM) were carried out. As expected, higher levels of methylation were observed for most H3 peptides under these conditions after 3 h (Fig. S27). Methylation of H3C1–C6benz (2) and H3triaz1-6 (3) peptides proceeded efficiently, reaching ∼86% and ∼95% after 3 h incubation, respectively (Fig. S27). The H3R2-E6 (12) and H3G0-E6 (13) peptides underwent ∼75% and ∼91% methylation by SETD7 (Fig. S27). In contrast, only traces (<5%) of methylation were observed for H3G1-E5 (14), while low level of methylation (∼11%) was observed for the H3G1-E7 (15) peptide (Fig. S27). The linear peptide H3G1E6 (16) was methylated to a higher degree, reaching ∼40% after 3 h (Fig. S27). Since many of the stapled peptides were fully methylated under initial conditions (500 nM SETD7, Fig. 2A), we also lowered the enzyme concentration to further distinguish the substrate efficiency between the best H3 substrates. MALDI-TOF MS-based enzymatic assays were carried out with 100 nM SETD7 in the presence of a stapled H3 peptide (10 µM) and SAM (30 µM) for 1 h at 37 °C (Fig. S28). The H3A1-E6 (5) peptide underwent ∼10% methylation, while the stereochemically inverted H3D-A1-E6 (6) appeared to be efficiently methylated by SETD7, showing ∼89% conversion (Fig. S28). SETD7 very efficiently catalysed methylation of H3G1-E6 (7), yielding ∼96% conversion after 1 h (Fig. S28). Substitution of Ala1 by Val (8) or Phe (9) resulted in ∼64% and ∼62% methylation, respectively, further demonstrating that bulkier side chains within the lactam-stapled peptide are also well tolerated by SETD7 (Fig. S28). In contrast, the stapled H3βA1-E6 (11) peptide underwent ∼48% methylation, suggesting that a shorter and more rigid ring (as in 7) is better accommodated by SETD7, in line with previous assays (Fig. S28).
After the screening of our panel of stapled H3 peptides, we carried out time-course experiments of SETD7-catalysed methylation to evaluate the substrate efficiencies (Fig. 2B and C). Incubation of the histone peptides (10 µM) with SETD7 (500 nM) and SAM (30 µM) showed a clear difference in SETD7's selectivity for the peptides, as reflected by the increase of methylation conversions over time (Fig. 2B). Eight of the stapled peptides (3, 5, 6, 7, 8, 9, 10 and 11) appeared to have faster reaction rates compared to the linear ‘native’ H3K4 (1) substrate (Fig. 2B). The H3A1-D6 (4) peptide, however, was a comparable substrate to the H3K4 substrate, whereas the H3C1–C6benz (2) peptide was observed to be a poorer SETD7 substrate than the H3K4 (1) reference. Peptides that appeared to be excellent substrates were further evaluated by incubation with 100 nM SETD7 and methylation conversions were detected at different time-points (Fig. 2C). Lowering the enzyme concentration resulted in the lack of H3K4 (1) methylation, but exceptional conversions of peptides 6 and 7, which underwent ∼90% methylation by SETD7 within 30 min (Fig. 2C). Notably, these two substrates appeared to outcompete any other stapled and linear substrate from our panel.
To further examine the catalytic efficiency of SETD7, we sought to determine kinetic parameters for the substrates that appeared to have considerably high turnover numbers in the presence of 100 nM SETD7 (3, 5, 6, 7, 8, 9 and 11) and compared them with the linear H3K4 (1) reference. Kinetics analyses were carried out under steady-state conditions by incubating SETD7 with varying concentrations of stapled H3 peptides (Table 1, Fig. 2D–F, S29 and Table 1). The H3W1-E6 (10) peptide was not evaluated since its turnover appeared to be low (Fig. 2C). The enzyme kinetics results demonstrated that peptides 3, 8 and 9 are a 7-fold, 5-fold and 6-fold better SETD7 substrates than H3K4 (1), respectively, as determined by increased kcat/Km values (Table 1 and Fig. S29), whereas peptides 5 and 11 are 10-fold and 12-fold better substrates when compared to the linear H3K4 (1) substrate (Table 1 and Fig. S29). The increase in kcat/Km values can be attributed to the more favourable Km values (Table 1). Notably, H3D-A1-E6 (6) and H3G1-E6 (7) peptides were found to display exceptional 97-fold and 110-fold increased kcat/Km values (1.33 µM−1 min−1 and 1.50 µM−1 min−1, respectively), which is a result of their high kcat values and low Km values (Fig. 2E, F and Table 1). The exceptionally low Km value observed for 7 can be attributed to the sterically smallest stapling linkage required for strong binding to SETD7. These results suggest that the amide-bond stapling between Gly1/D-Ala1 and Glu6 results in far superior SETD7 substrates when compared to the ‘native’ linear H3K4 peptide.
| Peptide | k cat (min−1) | K m (µM) | k cat/Km (µM−1 min−1) |
|---|---|---|---|
| 1 | 0.84 ± 0.08 | 61.6 ± 12.6 | 0.014 |
| 3 | 3.59 ± 0.39 | 36.2 ± 7.54 | 0.099 |
| 5 | 1.86 ± 0.14 | 12.5 ± 2.77 | 0.15 |
| 6 | 13.8 ± 0.84 | 10.4 ± 1.88 | 1.33 |
| 7 | 2.76 ± 0.17 | 1.84 ± 0.27 | 1.50 |
| 8 | 2.54 ± 0.26 | 34.1 ± 7.76 | 0.074 |
| 9 | 2.96 ± 0.31 | 34.6 ± 9.78 | 0.086 |
| 11 | 2.97 ± 0.19 | 17.7 ± 3.15 | 0.17 |
Having shown that SETD7 displays the highest substrate selectivity towards the stapled peptide 7, we evaluated the selectivity of other known and related SET-domain containing H3K4 lysine methyltransferases, MLL3 (KMT2C) and SETD1A (KMT2F), for this stapled peptide (Fig. 3). The methyltransferases MLL3 and SETD1A are catalytic subunits of COMPASS-complexes and structural homologues that predominantly catalyse H3K4 methylation at enhancers.42 Given this fact, we sought to assess whether these two enzymes do catalyse K4 methylation of stapled H3G1-E6 in a similar manner to SETD7. We synthesised longer linear H3K4 and stapled H3G1-E6 substrate peptides (residues 1–21), since the peptide length appears to be important for recognition and catalysis for the two enzymes. The SETD7-catalysed reaction of the 21-mer H3K4 substrate resulted in ∼88% methylation after 3 h, while the H3G1-E6 peptide showed ∼73% monomethylation and ∼25% dimethylation by SETD7, reaching ∼57% dimethylation after 24 h (Fig. 3A–C). This is an interesting finding, given that SETD7 is known to catalyse exclusive monomethylation of lysine residues in histone and non-histone substrates,15 however, we have recently observed that SETD7 has a capacity to catalyse partial dimethylation of high-affinity inhibitors.27 Human methyltransferase MLL3 displayed ∼73% monomethylation of the H3K4 substrate after 3 h and showed no activity for the stapled H3G1-E6 peptide within the detection limits even upon prolonged incubation (Fig. 3A–E). Furthermore, the human SETD1A-catalysed reaction of 21-mer H3K4 yielded ∼45% monomethylated product and ∼9% dimethylated product after 3 h, while no methylation was observed for the stapled H3G1-E6 after 24 h (Fig. 3A–G). These results suggest that the H3 peptide adopts different binding conformations in complex with MLL3 or SETD1A, demonstrating that the stapled H3G1-E6 peptide is highly selective for SETD7 over the related lysine methyltransferases MLL3 and SETD1A. While the MLL3-H3K4 complex structure suggests that positions 1 and 6 are not in close proximity,42 supporting the observation for lack of methylation of the stapled H3G1-E6 peptide, no biostructural information is available for H3 bound to SETD1A.
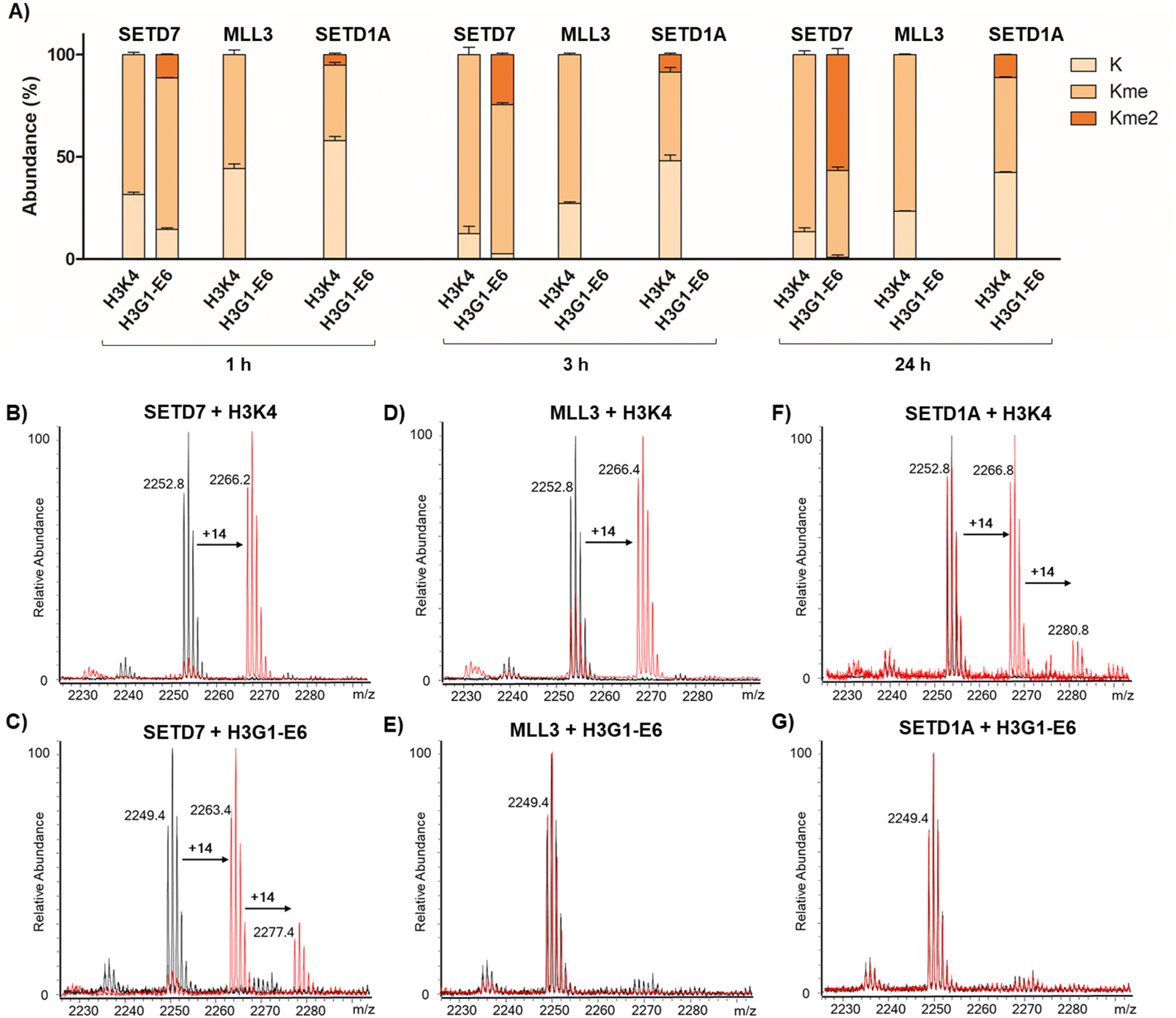 | ||
| Fig. 3 Selectivity assays of KMT-catalysed methylation of H3K4 and H3G1-E6 peptides (residues 1–21) after 1 h, 3 h and 24 h at 37 °C. Conversion of (A) SETD7-, MLL3-, and SETD1A-catalysed methylation of 21-mer linear and stapled H3 peptides at different time points. Error bars are reported as standard deviations from two independent replicates (n = 2). SETD7-catalysed (500 nM) methylation of (B) linear H3K4 (10 µM) and (C) stapled H3G1-E6 (10 µM) in the presence of SAM (30 µM). MLL3-catalysed (200 nM) methylation of (D) linear H3K4 (3 µM) and (E) stapled H3G1-E6 (3 µM) in the presence of SAM (100 µM). SETD1A-catalysed (1 µM) methylation of (F) linear H3K4 (2 µM) and (G) stapled H3G1-E6 (2 µM) in the presence of SAM (100 µM) (black: control reaction in absence of enzyme, red: enzymatic methylation reaction). | ||
Having demonstrated that the H3G1-E6 peptide is a superior substrate of SETD7, we sought to develop peptidomimetic inhibitors of SETD7 by substituting the K4 substrate residue with inactive methionine (Met), norvaline (Nva), norleucine (Nle) and 2-aminoheptanoic acid (Ahp) analogues as a part of the stapled 13-mer H3G1-E6 peptide (Fig. 4A). Histone lysine-to-methionine variants at different sites have been reported to reduce methylation levels (i.e., H3K4, H3K9 and H3K36 sites) and disrupt interactions between biomolecular partners involved in chromatin signaling pathways, often leading to cancer.43–45 For this purpose, we synthesised a longer 21-mer reference substrate peptide (H3K4, residues 1–21) for use as control in the MALDI-TOF MS-based inhibition assays. The synthetic stapled peptides (50 µM) were preincubated with human SETD7 (500 nM) and SAM (30 µM) for 10 min, after which the H3K4 substrate (residues 1–21, 10 µM) was added, and the reaction was carried out for additional 30 min at 37 °C. Initially, we investigated whether such peptides act as SETD7 inhibitors by employing single point (at 50 µM) assays and observed that SETD7 was ∼55% inhibited in the presence of the stapled peptide possessing Met (H3M4G1-E6, 17), while the linear counterpart 18 showed ∼31% inhibition (Fig. 4B). The stapled H3Nva4G1-E6 (19) and linear H3Nva4G1E6 (20) peptides displayed ∼47% and ∼33% inhibition, respectively (Fig. 4B). Notably, efficient SETD7 inhibition was observed with the stapled and linear H3Nle4 peptides 21 and 22, showing 100% and ∼77% inhibition, respectively (Fig. 4B). Remaining peptides (23 and 24) displayed ∼34% and ∼22% inhibitory activity against SETD7. The half-maximum inhibitory concentration (IC50) values were determined for peptides that showed good inhibition at 50 µM (17, 19, 21 and 22) (Fig. 4C). The stapled H3M4G1-E6 (17) peptide was found to have an IC50 of 56.5 µM, while H3Nva4G1-E6 (19) showed an IC50 of 83.1 µM (Fig. 4C). The most potent inhibition of human SETD7 was observed in the presence of the stapled H3Nle4G1-E6 (21) peptide (IC50 = 1.44 µM), whereas the linear H3Nle4G1E6 (22) peptide manifested a less pronounced inhibition (IC50 = 18.4 µM). We also demonstrated that the stapled 13-mer H3Nle4G1-E6 (21) peptide efficiently competes with the 13-mer H3K4 substrate, showing an IC50 value of 0.95 µM (Fig. S30). Substituting carbon-based analogues for K4 proved most effective with four-carbon side chain of Nle, while the shorter Nva and longer Ahp were less favourable, indicating that Nle possesses the optimal side-chain length for efficient inhibition of human SETD7. The stapled Met-peptide also displayed moderate inhibition, further suggesting that this side-chain length seems to be most suitable. Although the K-to-M variants are known to modulate histone methylation, the effects of H3K4M on H3K4 methylation vary and may depend on the system being studied.46 Overall, our results demonstrate that the inhibition of human SETD7 can be improved by appropriately stapling of histone H3 tail peptides.
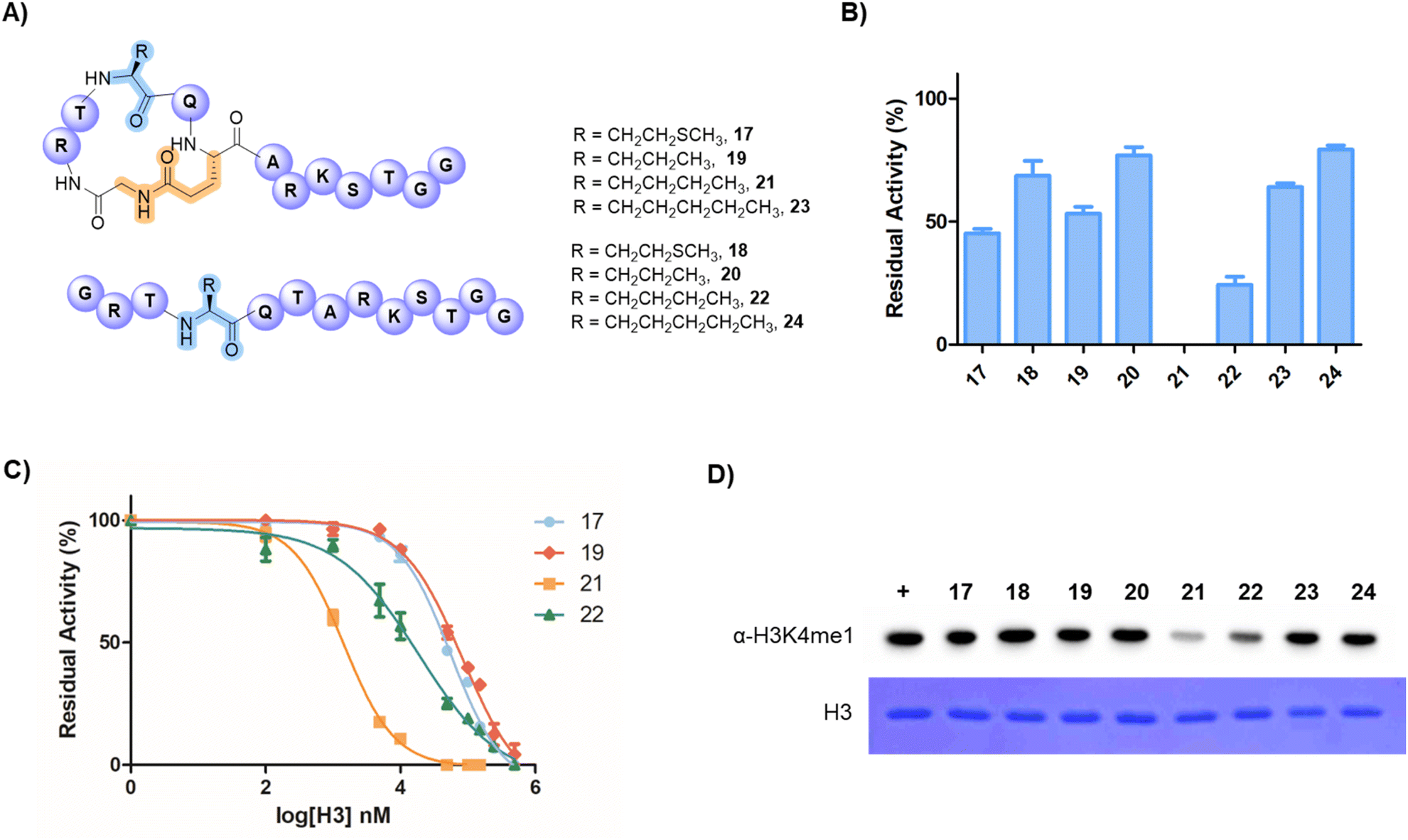 | ||
| Fig. 4 Inhibition assays of SETD7-catalysed methylation of H3K4. (A) Structures of selected stapled H3X4G1-E6 and their corresponding linear peptides. (B) Single point inhibition data of SETD7 (500 nM) by stapled/linear H3 peptides (50 µM) in the presence of the H3K4 substrate (residues 1–21, 10 µM) and SAM (30 µM). Error bars are reported as standard deviations from two independent replicates (n = 2). (C) Dose-response curves showing SETD7 inhibition by peptides 17, 19, 21 and 22. Error bars are reported as standard deviations from three independent replicates (n = 3). (D) Representative western blot analysis showing the detection of histone H3K4 monomethylation by SETD7 in the presence of stapled/linear H3 peptides (100 µM) and SAM (30 µM) (top), and Coomassie stain of total histone H3 protein loaded (bottom). | ||
We further evaluated the inhibition of SETD7-catalysed methylation of the full-length histone H3 in the presence of H3 peptides 17–24 by performing western blot experiments using the H3K4me1-specific antibodies (Fig. 4D and S31). SETD7 was preincubated with potential inhibitor peptide (100 µM) for 10 min in the presence of SAM (30 µM), followed by the incubation with full-length H3 protein (10 µM) for 30 min. The results demonstrate that the H3K4me1 levels are significantly reduced in the presence of peptides 21 and 22 relative to the control in the absence of inhibitors, resulting in ∼80% and ∼65% inhibition, respectively (Fig. 4D and S31). The results for peptide 21 align well with the data at the peptide level, although SETD7 appears to display higher preference for the full-length H3 protein. Taken together, the results demonstrate that the cyclic H3Nle4G1-E6 (21) peptide is the most potent peptide-based inhibitor of SETD7, and that inhibitory activity of the H3 peptides can be improved by 1,6-lactam stapling.
To investigate the expected effects of H3 cyclisation at the atomic level, we conducted two series of molecular dynamics (MD) simulations: one for the biostructurally obtained 10-mer linear H3K4 peptide and one for the cyclised/stapled variant H3K4G1-E6. Each series consisted of eight replicate simulations, each 100 ns in duration. We focused specifically on the interactions between Ala1 and Thr6 to assess whether a covalent linkage between these residues would disrupt the dynamics of the SETD7-H3 bound complex (Fig. 5A). To probe this, we computed a pairwise distance matrix of all heavy atoms between Ala1 and Thr6 (Fig. 5B and C). This analysis revealed that the side chain of Thr6 generally remains in proximity to Ala1 throughout the simulation, suggesting that a covalent linkage between these residues is sterically feasible. Further analysis of the distances between the hydroxyl oxygen of Thr6 and the nearest heavy atoms of Ala1 showed relatively stable separations, indicating that the crystal structure conformation is a representative snapshot of their typical spatial relationship (Fig. 5D).
 | ||
| Fig. 5 Molecular dynamics (MD) simulations of the SETD7-H3K4-SAH complexes. (A) Crystal structure of SETD7 (white) bound to a 10-mer H3 peptide fragment (blue) (obtained from PDB ID: 1O9S). Yellow dashed lines indicate hydrogen bonds; purple dashed lines represent salt bridges. (B) Amber atom names for heavy atoms of Ala1 and Thr6. (C) Heat map showing average interatomic distances between atoms identified in panel B. (D) Temporal distances between selected atoms during MD simulations for linear and stapled peptides. Solid lines represent average distances; shaded areas indicate standard error of the mean (SEM). (E) Root mean squared fluctuations (RMSF) of backbone atoms in the linear versus stapled peptide during MD simulations. (F) Root mean squared deviations (RMSD) of SAH with respect to the crystal structure (solid lines), and distance between the sulfur atom of SAH and the ε-amino group of LYS4 (dashed lines). Shaded areas denote SEM. | ||
Having confirmed the persistent proximity between Ala1 and Thr6 side chains, we next examined how cyclisation affects the H3 peptide conformation. Specifically, we measured the distance between the backbone atoms of residues 1 and 6 in both the linear and cyclised H3 peptides. Cyclisation led to a modest but consistent reduction and stabilisation of this inter-residue distance (6.6 ± 0.8 Å vs. 6.0 ± 0.3 Å; Fig. 5D). Interestingly, while the linear peptide fully unfolded in the unbound simulations (Ala1–Thr6 11.0 ± 2.4 Å), the cyclised peptide indeed maintained a folded conformation with stable inter-residue distances (6.2 ± 0.3 Å), supporting that cyclisation preorganises the peptide prior to binding (Fis. S32 and S33). One potential concern is that altering backbone geometry through cyclisation might destabilise the peptide's binding pose. However, root mean squared fluctuation (RMSF) analysis showed that the stapled H3 peptide maintained equal or greater stability across most residues compared to the linear H3 peptide (Fig. 5E). Notably, residues at the C-terminus (positions 7–10) appeared slightly more rigid in the cyclised form, likely due to torsional constraint introduced at residue 6 by the cyclisation.
Overall, the MD simulations suggest that cyclisation between residues 1 and 6 in H3 is well tolerated and may even enhance binding stability. As a final check, we monitored the distance between the ε-amino group of LYS4 and the sulfur atom of SAH, to ensure that cyclisation does not perturb this functional contact required for efficient methylation reaction. Consistent with experimental findings, we observed no significant changes in LYS4–SAH separation or in SAH binding (Fig. 5F). Taken together, these computational results support the experimental observations that cyclisation can preorganise the H3 tail peptide, reducing the entropic cost of binding while preserving its compatibility with SETD7 for efficient methyltransferase catalysis.
Conclusions
Our findings demonstrate that selected rationally designed stapled H3 peptide substrates can outcompete the linear H3K4 peptide substrate for lysine methyltransferase SETD7 catalysis. The enzymatic data reveal that most of the stapled H3 peptides are better substrates than the H3K4 substrate, indicating that the appropriately-constrained linking of the H3 peptide is favoured over the unstructured, linear counterpart found in nature. We demonstrate that the lactam-based 1,6-stapled H3 peptides are the most efficient SETD7 substrates when compared to those possessing the bis-thioether and triazole linkages. The observation that stapled H3G1-E5 and H3G1-E7 peptides were not good SETD7 substrates also highlights that the selectivity of SETD7 catalysis can be achieved by rationally designing the stapling sites in H3 peptides. Notably, the stapled H3G1-E6 and H3D-A1-E6 peptides display far superior catalytic efficiencies (110-fold and 97-fold increase) when compared to the linear H3K4 reference, making them SETD7 super-substrates. This finding is supported by the MD simulations, showing that the stapled H3G1-E6 peptide preorganises and enhances binding to SETD7. Inhibition assays reveal that the lysine-to-norleucine substituted H3G1-E6 peptide (H3Nle4G1-E6) acts as a potent, submicromolar, inhibitor of human SETD7. Considering the potential of peptide therapeutics for epigenetic proteins,47 which is mainly hampered due to the low membrane permeability and poor stability of linear peptides,48 there is currently a high need for development of chemical probes to address these challenges. Constrained macrocyclic/stapled peptides offer an advantage, as they display enhanced proteolytic and plasma stability relative to their linear counterparts and could display higher binding affinities.32,49–51 It is also no surprise that macrocyclic peptide-based modulators are gaining popularity in therapeutics, given their demonstrated high affinity and selectivity for targets, including epigenetic readers,52–56 writers57,58 and erasers.59,60 This work expands our knowledge on the SETD7 substrate binding and catalysis, and highlights the application of a proximity-guided design of stapled histone peptide fragments as peptide-based tool compounds with emerging biocatalytic and therapeutic potential, which will help the rational design of selective peptidomimetic modulators of SETD7, and by implication other epigenetic enzymes. We especially envision that with the availability of numerous X-ray structures of enzyme–histone complexes, it will be possible to rationally apply the proximity-guided approach for design and development of exceptionally efficient stapled super-substrates and highly potent peptide-based inhibitors of diverse histone-modifying enzymes (Fig. S34). More generally, the work demonstrates that peptide stapling should be considered as an appealing molecular approach in design of super-substrates that significantly outcompete the natural protein sequences.Experimental
Synthetic procedures
H3 peptides (residues 1–13, ARTKQTARKSTGG) were synthesised with C-terminal amides on Rink amide resin (0.78 g mmol−1 loading) employing automated Fmoc-SPPS chemistry on PurePep® Chorus Synthesizer. Coupling of amino acids was carried out by adding a mixture of amino acid (3.0 eq.) activated with hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) (3.0 eq.) and N,N-diisopropylethylamine (DIPEA) (5.0 eq.) in dimethylformamide (DMF) at 75 °C for 10 min; the coupling procedure was carried out twice for Arg. Fmoc deprotection was achieved by a solution of 20% (v/v) piperidine in DMF at 50 °C for 7 minutes. Elongations after the incorporation of the substituted Glu6 amino acid were either carried out via manual couplings or automated synthesis. Manual couplings were performed via standard couplings for 45 min and deprotections for 20 min at room temperature. After the final step in the synthesis, the peptides were washed with dichloromethane and dried over diethyl ether. Peptides proceeded to cleavage from resin using 95% (v/v) TFA, 2.5% (v/v) TIPS and 2.5% (v/v) Milli-Q water for 4 h. The crude peptides were precipitated with cold diethyl ether (−20 °C) and pelleted via centrifugation, purified by preparative reverse-phase HPLC, and lyophilized.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) and purified by RP-HPLC.
:2 v/v) and purified by RP-HPLC.
Enzyme assays
The initial screening for H3 substrate methylation by human SETD7 was performed in HEPES buffer (50 mM HEPES, 0.1 mM EDTA, 1 mM DTT, pH 8). H3 peptides (10 µM) were incubated with SAM (30 µM) and human SETD7 (100 nM, 500 nM or 1 µM) in a final volume of 30 µL. The reaction mixture was incubated at 37 °C while shaking at 750 rpm and quenched by the addition of 10% (v/v) TFA at different time points. Quenched samples were analysed by MALDI-TOF MS in duplicates (n = 2). Human MLL3 assays were performed by incubation of human MLL3 (200 nM), H3 peptide (3 µM) and SAM (100 µM) in reaction buffer (50 mM Tris, 2 mM MgCl2, 1 mM TCEP, pH 8.6) at 37 °C. Human SETD1A assays were performed by incubation of SETD1A (1 µM), H3 peptide (2 µM) and SAM (100 µM) in reaction buffer (50 mM Tris, 2 mM MgCl2, 1 mM TCEP, pH 8.6) at 37 °C. The % conversion of all methylation reactions was calculated from the ion chromatogram data by integrating peak areas using Flexanalysis software. Two independent enzyme assays experiments (n = 2) were carried out, with error bars reported as standard deviations. SETD7 enzyme kinetic assays were performed in a total reaction volume of 20 µL using a series of concentrations of the H3 peptide (1–150 µM) and SAM (30 µM). Reactions were initiated by the addition of SETD7 (50–300 nM) and incubated at 37 °C for 10 or 20 min while shaking. Quenched samples were analysed by MALDI-TOF MS from three independent assays. The % conversion of the methylation reaction was calculated from the ion chromatogram data by integrating peak areas using Flexanalysis software. Three independent enzyme kinetics experiments (n = 3) were carried out, with error bars reported as standard deviations.Inhibition assays
Single point screening assays were performed by incubating SETD7 (500 nM), H3 peptide variants (50 µM), and SAM (30 µM) for 10 min, followed by the addition of the 21-mer H3K4 substrate (10 µM), in a final volume of 20 µL in HEPES buffer (50 mM HEPES, 0.1 mM EDTA, 1 mM DTT, pH 8), and incubated for additional 30 min at 37 °C. IC50 measurements were carried out with H3 peptide variants (0.1 µM–500) and SAM (30 µM). Reactions were quenched by the addition of 10% (v/v) TFA and then analysed by MALDI-TIMS-TOF MS. The SETD7 activity was determined by calculating the integral of methylated peptide and normalised to a control reaction in absence of potential inhibitory peptides. Three independent inhibition assays (n = 3) were performed when measuring IC50 values, with error bars reported as standard deviations.Western blot assays
SETD7 inhibition assays were performed as described above with full-length H3 protein substrate (Sigma-Aldrich, #H2542); SETD7 (500 nM), SAM (30 µM) and H3 variants (100 µM). Reaction samples containing 1.7 µg histone H3 were loaded and separated on 15% bis–tris acrylamide gradient gel with Tris/SDS/glycine buffer, transferred to Nitrocellulose membrane with transfer buffer (40% (v/v) MeOH, 20% (v/v) Tris/glycine) and probed with primary antibody (Ab) (1:5000) for H3K4me1 (ThermoFischer Scientific, #703946) for 2 h. The membrane was blocked with SuperBlock blocking buffer (ThermoFischer Scientific) for 1 h before Ab incubation. After primary Ab incubation, the membrane was probed with anti-rabbit HRP secondary antibody (1:10000) for 1 h. The membrane was stained with the ECL substrate prior to detection and visualised with BioRad ChemiDoc imaging system. Three independent western blot experiments (n = 3) were carried out, with error bars reported as standard deviations.
MD simulations
The crystal structure of SETD7 bound to S-adenosylhomocysteine (SAH) and the H3K4me histone peptide fragment was obtained from the Protein Data Bank (PDB ID: 1O9S).15 The structure was imported into Maestro (Schrödinger Suite)61 for preparation and modification. LYS4 was manually demethylated, and protonation states were assigned using the protein preparation wizard. To construct the cyclised variant of the H3 peptide, Ala1 was mutated to glycine and Thr6 to glutamate. An amide bond was formed between the N-terminus of GLY1 and the side-chain carboxyl group of GLU6 using the Build panel in Maestro, followed by local energy minimisation. The resulting structures of SETD7, SAH, and both linear and cyclic peptides were then exported for simulation.For molecular dynamics (MD) simulations, the ff14SB force field was used to parameterise SETD7 and the H3 peptide fragments.62 SAH was treated using GAFF parameters.63 Partial atomic charges for SAH were derived via electrostatic potential (ESP) fitting using the RESP method in Antechamber,64 based on geometry-optimised structures from Gaussian09 at the HF/6-31G* level of theory.65 The cyclised GLU6 residue was parameterized analogously to SAH using the RESP approach. Prepgen was employed to generate a neutral charge state, and atom types were manually assigned to match the standard peptide backbone. For GLY1, standard charges and atom types from ff14SB were used.
System preparation for both the unbound H3 peptides and SETD7-H3 peptide complexes was performed using tleap.66 Each system was solvated in a TIP3P water box with a 12 Å buffer and 0.150 M NaCl. Eight replica simulations were run for each system. After initial energy minimisation (1000 steps), systems were heated to 300 K over 50 ps and equilibrated for 10 ns using the Berendsen barostat. Production simulations were carried out for 100 ns per replica. Trajectories from the production runs were analysed using the MDAnalysis package in Python.67
Author contributions
J. M. conceived and directed the study. N. B. carried out experiments under the supervision of J. M. L. M. performed molecular dynamics simulations under the supervision of J. K. N. B. and J. M. wrote the manuscript. All authors contributed to editing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: peptide synthesis, enzyme assays, inhibition assays, western blot assays, MD simulations. See DOI: https://doi.org/10.1039/d5sc08094k.Acknowledgements
This research was supported by the Lundbeck Foundation (R344-2020-1051 to J. M.) and the Novo Nordisk Foundation (NNF23OC0083407 to J. M.).References
- T. Kouzarides, Cell, 2007, 128, 693–705 CrossRef CAS PubMed.
- A. J. Andrews and K. Luger, Annu. Rev. Biophys., 2011, 40, 99–117 CrossRef CAS PubMed.
- K. Luger, M. L. Dechassa and D. J. Tremethick, Nat. Rev. Mol. Cell Biol., 2012, 13, 436–447 CrossRef CAS PubMed.
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381–395 CrossRef CAS PubMed.
- E. M. Cornett, B. M. Dickson, K. Krajewski, N. Spellmon, A. Umstead, R. M. Vaughan, K. M. Shaw, P. P. Versluis, M. W. Cowles, J. Brunzelle, Z. Yang, I. E. Vega, Z. W. Sun and S. B. Rothbart, Sci. Adv., 2018, 4, eaav2623 CrossRef CAS PubMed.
- K. Hyun, J. Jeon, K. Park and J. Kim, Exp. Mol. Med., 2017, 49, e324 CrossRef CAS PubMed.
- M. N. Maas, J. C. J. Hintzen and J. Mecinović, Chem. Commun., 2022, 58, 7216–7231 CAS.
- M. Luo, Chem. Rev., 2018, 118, 6656–6705 CrossRef CAS PubMed.
- M. M. Müller and T. W. Muir, Chem. Rev., 2015, 115, 2296–2349 Search PubMed.
- C. D. Allis and T. Jenuwein, Nat. Rev. Genet., 2016, 17, 487–500 CAS.
- J. C. Black, C. Van Rechem and J. R. Whetstine, Mol. Cell, 2012, 48, 491–507 CAS.
- E. L. Greer and Y. Shi, Nat. Rev. Genet., 2012, 13, 343–357 CAS.
- H. Wang, R. Cao, L. Xia, H. Erdjument-Bromage, C. Borchers, P. Tempst and Y. Zhang, Mol. Cell, 2001, 8, 1207–1217 CAS.
- K. Nishioka, S. Chuikov, K. Sarma, H. Erdjument-Bromage, C. D. Allis, P. Tempst and D. Reinberg, Gend. Dev., 2002, 16, 479–489 CAS.
- B. Xiao, C. Jing, J. R. Wilson, P. A. Walker, N. Vasisht, G. Kelly, S. Howell, I. A. Taylor, G. M. Blackburn and S. J. Gamblin, Nature, 2003, 421, 652–656 CAS.
- A. Shilatifard, Annu. Rev. Biochem., 2012, 81, 65–95 CAS.
- H. Wang and K. Helin, Trends Cell Biol., 2025, 35, 115–128 CAS.
- G. Gao, J. Dornelas Moreira, P. Das, K. Lin, K. Ge, T. Chen, Y. Lu and M. A. Santos, Nat. Commun., 2025, 16, 5737 Search PubMed.
- J.-H. Lee and D. G. Skalnik, Mol. Cell. Biol., 2008, 28, 609–618 CAS.
- J. Sugeedha, J. Gautam and S. Tyagi, Epigenetics, 2021, 16, 469–487 Search PubMed.
- M. J. Oudhoff, M. J. S. Braam, S. A. Freeman, D. Wong, D. G. Rattray, J. Wang, F. Antignano, K. Snyder, I. Refaeli, M. R. Hughes, K. M. McNagny, M. R. Gold, C. H. Arrowsmith, T. Sato, F. M. V. Rossi, J. H. Tatlock, D. R. Owen, P. J. Brown and C. Zaph, Dev. Cell, 2016, 37, 47–57 CrossRef CAS PubMed.
- R. Huang, X. Li, Y. Yu, L. Ma, S. Liu, X. Zong and Q. Zheng, Oncotarget, 2017, 8, 94080–94090 CrossRef PubMed.
- L. Gaughan, J. Stockley, N. Wang, S. R. C. McCracken, A. Treumann, K. Armstrong, F. Shaheen, K. Watt, I. J. McEwan, C. Wang, R. G. Pestell and C. N. Robson, Nucleic Acids Res., 2011, 39, 1266–1279 CrossRef CAS PubMed.
- A. Kouskouti, E. Scheer, A. Staub, L. Tora and I. Talianidis, Mol. Cell, 2004, 14, 175–182 CrossRef CAS PubMed.
- S. Chuikov, J. K. Kurash, J. R. Wilson, B. Xiao, N. Justin, G. S. Ivanov, K. McKinney, P. Tempst, C. Prives, S. J. Gamblin, N. A. Barlev and D. Reinberg, Nature, 2004, 432, 353–360 CrossRef CAS PubMed.
- X. D. Yang, B. Huang, M. Li, A. Lamb, N. L. Kelleher and L. F. Chen, EMBO J., 2009, 28, 1055–1066 CrossRef CAS PubMed.
- M. R. B. Porzberg, D. C. Lenstra, E. Damen, R. H. Blaauw, F. P. J. T. Rutjes, A. Wegert and J. Mecinović, ChemMedChem, 2023, 18, e202300457 CrossRef CAS PubMed.
- D. C. Lenstra, E. Damen, R. G. G. Leenders, R. H. Blaauw, F. P. J. T. Rutjes, A. Wegert and J. Mecinović, ChemMedChem, 2018, 13, 1405–1413 CrossRef CAS PubMed.
- D. Barsyte-Lovejoy, F. Li, M. J. Oudhoff, J. H. Tatlock, A. Dong, H. Zeng, H. Wu, S. A. Freeman, M. Schapira, G. A. Senisterra, E. Kuznetsova, R. Marcellus, A. Allali-Hassani, S. Kennedy, J.-P. Lambert, A. L. Couzens, A. Aman, A.-C. Gingras, R. Al-Awar, P. V. Fish, B. S. Gerstenberger, L. Roberts, C. L. Benn, R. L. Grimley, M. J. S. Braam, F. M. V. Rossi, M. Sudol, P. J. Brown, M. E. Bunnage, D. R. Owen, C. Zaph, M. Vedadi and C. H. Arrowsmith, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12853–12858 CrossRef CAS PubMed.
- H.-B. Guo and H. Guo, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8797–8802 CrossRef CAS PubMed.
- L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol., 2017, 48, 242–250 CrossRef CAS PubMed.
- X. Ji, A. L. Nielsen and C. Heinis, Angew. Chem., Int. Ed., 2024, 63, e202308251 CrossRef CAS PubMed.
- Y. Tanaka, C. J. Hipolito, A. D. Maturana, K. Ito, T. Kuroda, T. Higuchi, T. Katoh, H. E. Kato, M. Hattori, K. Kumazaki, T. Tsukazaki, R. Ishitani, H. Suga and O. Nureki, Nature, 2013, 496, 247–251 CrossRef CAS PubMed.
- K. Ito, K. Sakai, Y. Suzuki, N. Ozawa, T. Hatta, T. Natsume, K. Matsumoto and H. Suga, Nat. Commun., 2015, 6, 6373 CrossRef CAS PubMed.
- K. Yamagata, Y. Goto, H. Nishimasu, J. Morimoto, R. Ishitani, N. Dohmae, N. Takeda, R. Nagai, I. Komuro, H. Suga and O. Nureki, Structure, 2014, 22, 345–352 CrossRef CAS PubMed.
- M. K. Schuhmacher, S. Beldar, M. S. Khella, A. Bröhm, J. Ludwig, W. Tempel, S. Weirich, J. Min and A. Jeltsch, Commun. Biol., 2020, 3, 511 CrossRef CAS PubMed.
- P. Schnee, M. Choudalakis, S. Weirich, M. S. Khella, H. Carvalho, J. Pleiss and A. Jeltsch, Commun. Chem., 2022, 5, 139 CrossRef CAS PubMed.
- S. Weirich, D. Kusevic, P. Schnee, J. Reiter, J. Pleiss and A. Jeltsch, Commun. Biol., 2024, 7, 707 CrossRef CAS PubMed.
- D. P. Fairlie and A. Dantas de Araujo, Biopolymers, 2016, 106, 843–852 CrossRef CAS PubMed.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011–5014 CrossRef CAS PubMed.
- P. Grieco, P. M. Gitu and V. J. Hruby, J. Pept. Res., 2001, 57, 250–256 CrossRef CAS PubMed.
- Y. Li, L. Zhao, Y. Zhang, P. Wu, Y. Xu, J. Mencius, Y. Zheng, X. Wang, W. Xu, N. Huang, X. Ye, M. Lei, P. Shi, C. Tian, C. Peng, G. Li, Z. Liu, S. Quan and Y. Chen, Mol. Cell, 2022, 82, 3810–3825 CrossRef CAS PubMed.
- H.-M. Herz, M. Morgan, X. Gao, J. Jackson, R. Rickels, S. K. Swanson, L. Florens, M. P. Washburn, J. C. Eissenberg and A. Shilatifard, Science, 2014, 345, 1065–1070 CrossRef CAS PubMed.
- Y. Jang, A. Broun, C. Wang, Y.-K. Park, L. Zhuang, J.-E. Lee, E. Froimchuk, C. Liu and K. Ge, Nucleic Acids Res., 2018, 47, 607–620 CrossRef PubMed.
- K. M. Chan, J. Han, D. Fang, H. Gan and Z. Zhang, Cell Cycle, 2013, 12, 2546–2552 CrossRef CAS PubMed.
- K. Serdyukova, A. R. Swearingen, M. Coradin, M. Nevo, H. Tran, E. Bajric and J. Brumbaugh, Development, 2023, 150, dev202169 CrossRef CAS PubMed.
- J. C. J. Hintzen and J. Mecinović, in Progress in Nucleic Acid Research and Molecular Biology, Academic Press, 2025, 212, pp. 25–65 Search PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discov., 2021, 20, 309–325 CrossRef CAS PubMed.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed.
- X. Li, S. Chen, W. D. Zhang and H. G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
- L. Lombardi, V. D. Genio, F. Albericio and D. R. Williams, Chem. Rev., 2025, 125, 7099–7166 CrossRef CAS PubMed.
- C. A. Hurd, J. T. Bush, A. J. Powell and L. J. Walport, Angew. Chem., Int. Ed., 2024, 63, e202406414 CrossRef CAS PubMed.
- P. Betlem, M. N. Maas, J. Middelburg, B. J. Pieters and J. Mecinović, Chem. Commun., 2022, 58, 12196–12199 RSC.
- M. Y. Zhang, H. Yang, G. Ortiz, M. J. Trnka, N. Petronikolou, A. L. Burlingame, W. F. DeGrado and D. G. Fujimori, Chem. Sci., 2022, 13, 6599–6609 RSC.
- O. D. Coleman, J. Macdonald, B. Thomson, J. A. Ward, C. J. Stubbs, T. E. McAllister, S. Clark, S. Amin, Y. Cao, M. I. Abboud, Y. Zhang, H. Sanganee, K. V. M. Huber, T. D. W. Claridge and A. Kawamura, Chem. Sci., 2023, 14, 7136–7146 RSC.
- K. Patel, L. J. Walport, J. L. Walshe, P. D. Solomon, J. K. K. Low, D. H. Tran, K. S. Mouradian, A. P. G. Silva, L. Wilkinson-White, A. Norman, C. Franck, J. M. Matthews, J. M. Guss, R. J. Payne, T. Passioura, H. Suga and J. P. Mackay, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26728–26738 CrossRef CAS PubMed.
- M. T. Bertran, R. Walmsley, T. Cummings, I. V. Aramburu, D. J. Benton, R. Mora Molina, J. Assalaarachchi, M. Chasampalioti, T. Swanton, D. Joshi, S. Federico, H. Okkenhaug, L. Yu, D. Oxley, S. Walker, V. Papayannopoulos, H. Suga, M. A. Christophorou and L. J. Walport, Nat. Commun., 2024, 15, 9746 CrossRef CAS PubMed.
- I. R. Mathiesen, E. D. D. Calder, S. Kunzelmann and L. J. Walport, Commun. Chem., 2024, 7, 304 CrossRef CAS PubMed.
- A. Kawamura, M. Münzel, T. Kojima, C. Yapp, B. Bhushan, Y. Goto, A. Tumber, T. Katoh, O. N. F. King, T. Passioura, L. J. Walport, S. B. Hatch, S. Madden, S. Müller, P. E. Brennan, R. Chowdhury, R. J. Hopkinson, H. Suga and C. J. Schofield, Nat. Commun., 2017, 8, 14773 CrossRef CAS PubMed.
- D. Danková, A. L. Nielsen, A. Zarda, T. N. Hansen, M. Hesse, M. Benová, A. Tsiris, C. R. O. Bartling, E. J. Will, K. Strømgaard, C. Moreno-Yruela, C. Heinis and C. A. Olsen, J. Am. Chem. Soc., 2025, 5, 1299–1307 Search PubMed.
- Maestro, Schrödinger, LLC, New York, NY, 2020 Search PubMed.
- A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dunbrack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo and S. Ha, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden and R. E. Duke, Amber 2021, University of California, San Francisco, 2021 Search PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |