
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
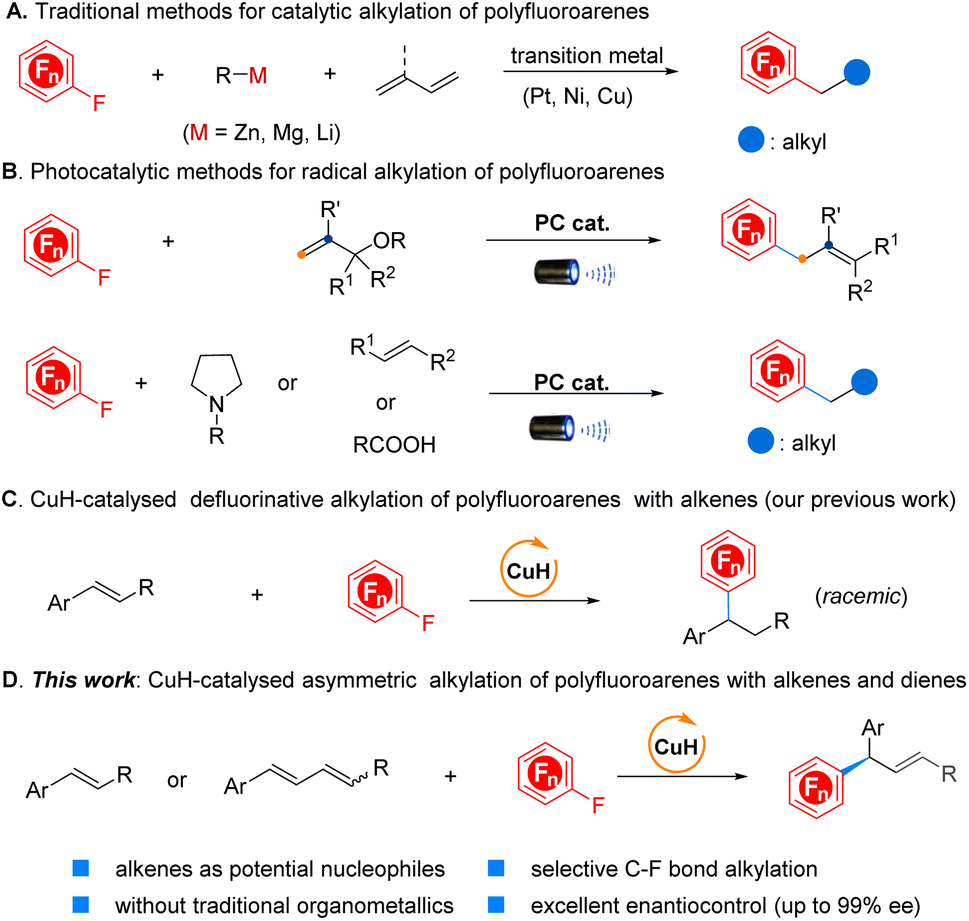
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective copper-catalysed defluorinative alkylation of polyfluoroarenes with alkenes and 1,3-dienes
Dazhen
Shi†
a,
Lihan
Zhu†
 a,
Ying
Jiang
a,
Simin
Wang
a,
Jianjun
Yin
a,
Xiuping
Yuan
a,
Shucheng
Ma
a,
Xiaoyu
Li
a,
Jiaqiong
Sun
a,
Qian
Zhang
*ab and
Tao
Xiong
*a
a,
Ying
Jiang
a,
Simin
Wang
a,
Jianjun
Yin
a,
Xiuping
Yuan
a,
Shucheng
Ma
a,
Xiaoyu
Li
a,
Jiaqiong
Sun
a,
Qian
Zhang
*ab and
Tao
Xiong
*a
aJilin Province Key Laboratory of Organic Functional Molecular Design & Synthesis, Department of Chemistry, Northeast Normal University, Changchun 130024, China. E-mail: xiongt626@nenu.edu.cn; zhangq651@nenu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China
First published on 25th November 2025
Abstract
Despite impressive advances in the field of defluorinative alkylation of polyfluoroarenes, the corresponding asymmetric counterpart has remained a formidable challenge. Herein, we present the first example of asymmetric defluorinative alkylation of polyfluoroarenes with readily accessible alkenes, 1,3-dienes, and even (Z/E)-mixed 1,3-dienes as potential nucleophiles in the presence of a chiral CuH catalyst, avoiding the conventional utilization of highly reactive organometallics. This method enables the efficient construction of the challenging chiral Csp3–CArF bond with outstanding stereocontrol (up to 99% ee in most cases). This reaction proceeds under mild reaction conditions, demonstrating excellent functional group compatibility and high regio- and stereoselectivity. Experimental studies in conjunction with density functional theory (DFT) calculations were carried out to unravel the plausible mechanism and elucidate the origins of excellent enantioselectivity.
Introduction
The development of methods for the selective incorporation of polyfluorinated aromatic motifs into organic molecules has garnered significant attention. This interest stems from the exceptional properties exhibited by these compounds, including enhanced lipophilicity, metabolic stability, improved passive diffusion of compounds across membranes, and the low oxidation potential, which have been extensively utilized in pharmaceutical chemistry, agricultural chemistry, and materials science (Fig. 1).1 Despite the great value, the synthesis of such compounds has remained largely reliant on harsh methods, exemplified by the Balz–Schiemann decomposition of aryl diazonium tetrafluoroborate salts2 or the Halex (over 200 °C) process.3 Recently, transition-metal-catalysed fluorination of prefunctionalized arenes and directed C–H fluorination have emerged as promising strategies for approaching fluorinated arenes;4 however, these methods are not well suitable for the preparation of polyfluorinated arenes, due to the need for multiple prefunctionalizations or specifically arranged directing groups. By comparison, the functionalization of readily available and inexpensive polyfluoroarenes via site selective C–F bond activation might represent an efficient complementary approach to overcome these existing limitations, offering a more general and versatile route to sculpt far more complex fluorinated arenes that have historically been synthetically inaccessible. In this regard, notable progress has been made regarding transition-metal-catalysed hydrodefluorination (HDF),5 C–F bond borylation,6 and C–F bond arylation7 in the last few years. Nevertheless, the more challenging selectively defluorinative Csp2–Csp3 bond formation, particularly in an enantioselective manner, remains an underdeveloped area. | ||
| Fig. 1 Selected examples of important polyfluoroaryl compounds. | ||
The recently emerging “escape from flatland” concept, which potentially enhances the likelihood of clinical success for candidate drug molecules, has significantly spurred the exploitation of new approaches for C(sp3) centre creation.8 However, the corresponding progress in selective C–F bond alkylation of polyfluoroarenes to construct the CArF–Csp3 bond remains extremely sluggish. This situation is likely attributed to the high C–F bond dissociation energy (BDE = 145 kcal mol−1 in C6F6),9 poor site selectivity control, the tendency of undesirable β-H elimination and the formation of the thermodynamically stable M–F bond (M: transition metal) which hinders catalyst turnover. To alleviate these challenges, early efforts primarily focused on using highly reactive stoichiometric alkylmetallic reagents as coupling partners,10 while the intrinsic drawbacks of organometallics impaired the functional group tolerance and synthetic efficiency (Scheme 1A).
 | ||
| Scheme 1 Catalytic defluorinative alkylation of polyfluoroarenes. | ||
Inspired by the recent advancements in photoredox chemistry enabling the generation of highly reactive radical species under mild conditions, Weaver and co-workers have ingeniously achieved a series of addition reactions of in situ-formed polyfluorinated arene radical species to unsaturated bonds, offering an efficient approach for constructing the CArF–Csp3 bond.11 Alternatively, Hashmi's group developed a promising visible-light-induced defluoroalkylation via radical–radical coupling (Scheme 1B, top).12 Quite recently, Ritter, Xu, Wu and other groups have well demonstrated the feasibility of nucleophilic alkyl radical addition to polyfluoroarene as the critical step with photocatalysis for preparing alkylated polyfluoroarenes (Scheme 1B, bottom).13 Additionally, Hu's laboratory established an alternative route for CArF–Csp3 bond formation through the decarboxylative coupling reaction of aliphatic N-hydroxyphthalimide esters with polyfluoroaryl zinc reagents with synergetic photoredox and copper catalysis.14 Despite the unique advantages of each strategy, achieving enantioselective defluorinative alkylation still poses a permanent challenge.15 On the other hand, the development of stereoconvergent asymmetric reactions with (Z/E)-mixed alkenes has been identified as a highly sustainable and emerging approach for transforming mixed stereoisomers into an optically pure product. Conventional olefination processes such as the Wittig reaction, olefin metathesis reaction, and Horner–Wadsworth–Emmons (HWE) reaction, among others or those produced directly from petroleum cracking, typically provide an isomeric alkene mixture. However, in contrast to this circumstance, the stereochemical information on alkenes such as a (Z)- or (E)-configuration still plays a critical role in determining the absolute configuration of the resulting chiral product within the currently available arsenal of the synthetic tool kit; otherwise, it will compromise the yield and stereoselectivity if an alkene is a mixture of (Z/E)-isomers. As a consequence, it is essential to carry out the difficult and time-consuming purification of the alkenes to ensure the high stereopurity of the final products. Despite the significant interest, developing effective stereoconvergent and asymmetric methods has been proven difficult and only a few examples have been reported to date with encompassing transition-metal catalysis and emerging biocatalysis,16 particularly in simultaneously achieving high enantioselectivity and the geometrical configuration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the resulting products.
C bond in the resulting products.
Recent advances in CuH chemistry have highlighted the merits of using bench-stable unsaturated hydrocarbons as alternatives to stoichiometric organometallics in coupling reactions.17 Accordingly, considerable efforts have been exerted to develop new hydrofunctionalization reactions, however, only sporadic asymmetric hydro(hetero)arylation reactions of alkenes and 1,3-enynes with aryl bromides, pyridines, pyridazines, quinoline N-oxides or 2-fluoropyridine have been reported by the groups of Buchwald, Ge, and Kim, respectively.18 In 2020, our group disclosed the first example of defluorinative alkylation of polyfluoroarenes with alkenes by CuH catalysis, nevertheless, the corresponding asymmetric transformation has remained to be conquered (Scheme 1C).19 Due to our continuing interest in CuH catalysis20 and recent stereoconvergent asymmetric conversion,21 we herein describe the first CuH-catalysed asymmetric defluorinative alkylation of polyfluoroarenes with alkenes and 1,3-dienes. Furthermore, the corresponding transformations with a mixture of (Z/E)-1,3-dienes are also realized, stereoconvergently affording a series of multidiscipline-related yet difficult-to-access chiral polyfluoroarenes in high efficiency with excellent enantioselectivity (Scheme 1D).
Results and discussion
Optimization of reaction conditions
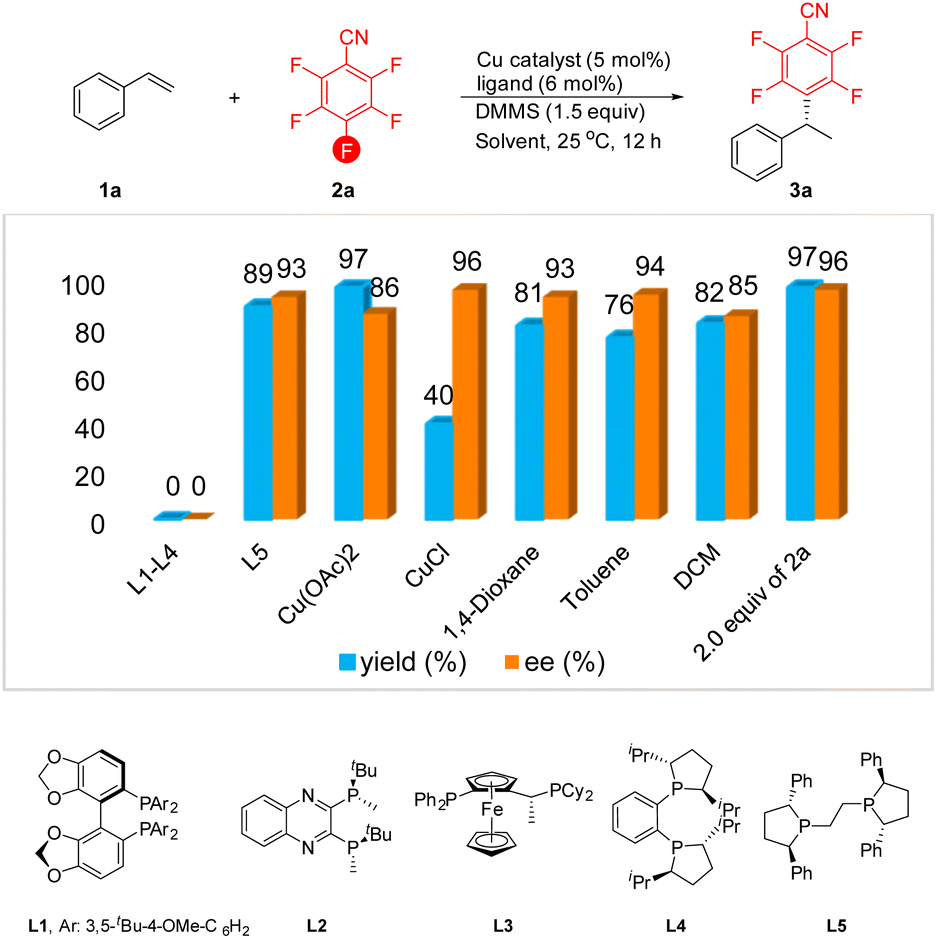
We initiated our investigation with styrene 1a and commercially available pentafluorobenzonitrile 2a as the model substrates. In the preliminary stages of our attempts to probe the feasibility of this conception, various chiral bidentate phosphine ligands were evaluated (Table 1). Unfortunately, several bidentate chiral diphosphine ligands failed to furnish the desired product, including DTBM-SegPhos L1, QuinoxP L2, Josiphos L3 and DuPhos L4. To our delight, the use of (S,S)-Ph-BPE L5 provided the anticipated product 3a in excellent yield with a high level of enantioselectivity. Further screening of copper salts revealed that Cu(OAc)2 was effective in improving the efficiency of the reaction but showed a relatively low level of enantiocontrol. Conversely, conducting the alkylation with CuCl as the catalyst led to a higher level of enantioselectivity, but the yield of 3a was reduced significantly. Other solvents such as 1,4-dioxane, toluene, and DCM were also investigated. Although the enantioselectivity was maintained at a good to excellent level, the yield of the product was slightly decreased. To our delight, when we increased the amount of styrene 1a to 2.0 equiv., the expected defluorinative product 3a was formed in 97% yield with 96% ee.| a Reaction conditions: 1a (0.3 mmol, 1.5 equiv.), 2a (0.2 mmol), DMMS (0.3 mmol, 1.5 equiv.), catalyst (5 mol%), ligand (6 mol%) in dry solvent (2.0 mL) at 25 °C for 12 h. The yield of 3a was determined via1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard, and ee values were determined by HPLC analysis. |
|---|
|
Substrate scope
With the optimized reaction conditions in hand, we assessed the functional group tolerance and scope of styrenes in this asymmetric defluorinative alkylation. As shown in Table 2, a wide range of styrenes containing either electron-donating (3a–3w) or electron-withdrawing (3x–3ae) groups on the aryl rings were readily accommodated. Ortho-, meta-, and para-methyl substituted styrenes (1b–1d) were converted to the desired products 3b–3d in good to excellent yields with 78–99% ee, despite the relatively poor yield and enantiocontrol for 3b, probably owing to the crowded steric hindrance. Further investigations revealed that a wide range of functional groups, such as alkyl (3e–3g), alkoxyl (3h–3j), phenoxyl (3k), silylether (3l), acetyl (3m), alkyl chloride (3n–3o), thioether (3s), amine (3t), pinacolborato (3u), halide (3x–3aa), trifluoromethyl (3ab), trifluoromethoxy (3ac), ester (3ad), and even ketone (3ae), were well tolerated under standard reaction conditions. In addition, unactivated alkenyl, internal alkynyl, and internal alkenyl groups were used to examine the chemoselectivity of this method, which showed that the alkylation occurs on the activated alkenyl group (3p–3r). These results reflected the reactive inertness of these functional groups. The highly reactive 2-vinylnaphthalene underwent the reaction smoothly to afford the corresponding product 3af in 87% yield with 99% ee. Moreover, several alkenes containing pharmaceutically valuable heterocycles proved to be competent substrates, allowing for the generation of products 3ag–3ai in moderate to excellent yields with up to >99% ee. In addition, the absolute configuration of 3y was determined by single-crystal X-ray diffraction analysis and the absolute stereochemistry of the other products was assigned by analogy. We next investigated the scope of polyfluoroarenes. In addition to the cyano group, both ester and trifluoromethyl-substituted pentafluorobenzenes were suitable for this protocol, providing the corresponding products 3aj–3ao in moderate to good yields with excellent enantioselectivity. Polyfluoropyridines were efficiently transformed into the alkylation products 3ap and 3aq in 79% and 82% yields with 92% and 93% ee, respectively. To our delight, polyfluoropyrimidine, which is widely available in biologically active molecules, also reacted to furnish 3ar in 57% yield with slightly low ee (73%).| a Reaction conditions: 1 (0.4 mmol, 2.0 equiv.), 2 (0.2 mmol), DMMS (0.3 mmol, 1.5 equiv.), Cu(C8H15O2)2 (5 mol%), (S,S)-Ph-BPE (6 mol%) in dry THF (2.0 mL) at 25 °C for 12 h. The yield of 3a was determined via1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard, and the ee value was determined by HPLC analysis. b The reactions were performed at −10 °C. c The reactions were performed for 36 h. |
|---|

|
Intrigued by these excellent results, we were next particularly interested in finding whether this method would be suitable for the more challenging defluorinative alkylation with 1,3-dienes as the coupling partner, which would construct a Csp3–CArF stereocentre and simultaneously introduce a synthetically versatile C–C double bond. However, in addition to the enantioselectivity control, the additional challenges, including regioselectivity (1,4- versus 3,4-addition), and stereochemistry of the resulting C–C double bond remain to be addressed. To our delight, the targeted chiral 1,4-Markovnikov products 5a can be efficiently obtained with almost perfect enantioselectivity (>99% ee) and exclusive trans-configuration of the C–C double bond through the slight modification of the optimal conditions (Table 3). The steric effects on the phenyl rings in alkenes were almost negligible in this reaction, demonstrated by 5b–5d and 5h–5j. In addition, either electron-donating or withdrawing groups with various useful functionalities such as alkyl, ether, amine, phenyl, halogens, and trifluoromethyl on the aromatic rings were all readily accommodated, providing products 5b–5o in constantly excellent yields (73–98%) and ee values (93–>99%). Stereoconvergent transformation of (Z/E)-mixed alkenes into a single chiral product is an extremely attractive strategy, owing to the avoidance of the difficult and time-consuming purification of the alkene mixture. To our delight, this catalytic system could also be applicable to (Z/E)-mixed dienes, which provided 5p–5t stereoconvergently in the range of 80–91% yield with 98–>99% ee. Likewise, polyfluoroarenes with different functionalities also showed excellent reactivity with either stereodefined 1,3-dienes or (Z/E)-mixed 1,3-dienes as the starting materials, giving rise to chiral products 5u–5aa with generally not less than 99% ee. The relatively low efficiency observed for 5w was attributed to the generation of a large amount of the unproductive defluorinative hydrogenation product, accompanied by the recovery of 46% yield of the 1,3-diene starting material. Notably, almost unchanged Z- and E-alkene isomeric ratios of the recovered (Z/E)-1,3-diene mixture were observed in these reactions, implying that the configuration of the alkene had no prominent effect on reactivity. In addition to styrenes and 1,3-dienes, we also evaluated whether 1,1-disubstituted alkenes or internal alkenes, such as α- or β-alkyl substituted styrenes, were compatible with this approach, however, no desired coupling product was detected using the current catalytic system. Besides, the less electrophilic polyfluoroarenes bearing electron-donating substituents such as alkyl or alkoxy functional groups were also inert for this transformation. To further demonstrate the robustness of this protocol, we subsequently evaluated a series of alkene containing molecules derived from architecturally complex scaffolds—including diacetonefructose, estrone, vitamin E, ibuprofen, menthol, borneol, and cholesterol—featuring potentially vulnerable functional groups such as ketone, ester and ketal. The corresponding products 6–13 were obtained in moderate to good yields (47–95%) with excellent diastereoselectivities (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.), thereby demonstrating the compatibility and potential of this protocol for late-stage diversification of complex bioactive molecules.
:1 d.r.), thereby demonstrating the compatibility and potential of this protocol for late-stage diversification of complex bioactive molecules.
| a Reaction conditions: 4 (0.4 mmol, 2.0 equiv.), 2b (0.2 mmol), PhSiH3 (0.3 mmol, 1.5 equiv.), Cu(C8H15O2)2 (5 mol%), (S,S)-Ph-BPE (6 mol%) in dry THF (2.0 mL) at 0 °C for 6 h. The yield of 5 was determined via1H NMR analysis of the crude reaction mixture using CH2Br2 as an internal standard, and the ee value was determined by HPLC analysis. b The reactions were performed with (TMSO)2MeSiH (0.6 mmol, 3.0 equiv.) at 25 °C for 12 h. c The reactions were performed for 24 h. d The reactions were performed with DMMS (0.3 mmol, 1.5 equiv.) at 25 °C for 12 h. e The reactions were performed at 30 °C. |
|---|

|
Mechanistic investigation
A series of mechanistic experiments were then conducted to gain deeper insights into this copper-catalysed asymmetric defluorinative alkylation of polyfluoroarenes. As depicted in Fig. 2A, the defluorinative alkylation between substrates 1a and 2a was significantly suppressed, with a substantial amount of ethylbenzene (14) being generated, when a stoichiometric amount of iPrOH was added under optimal reaction conditions.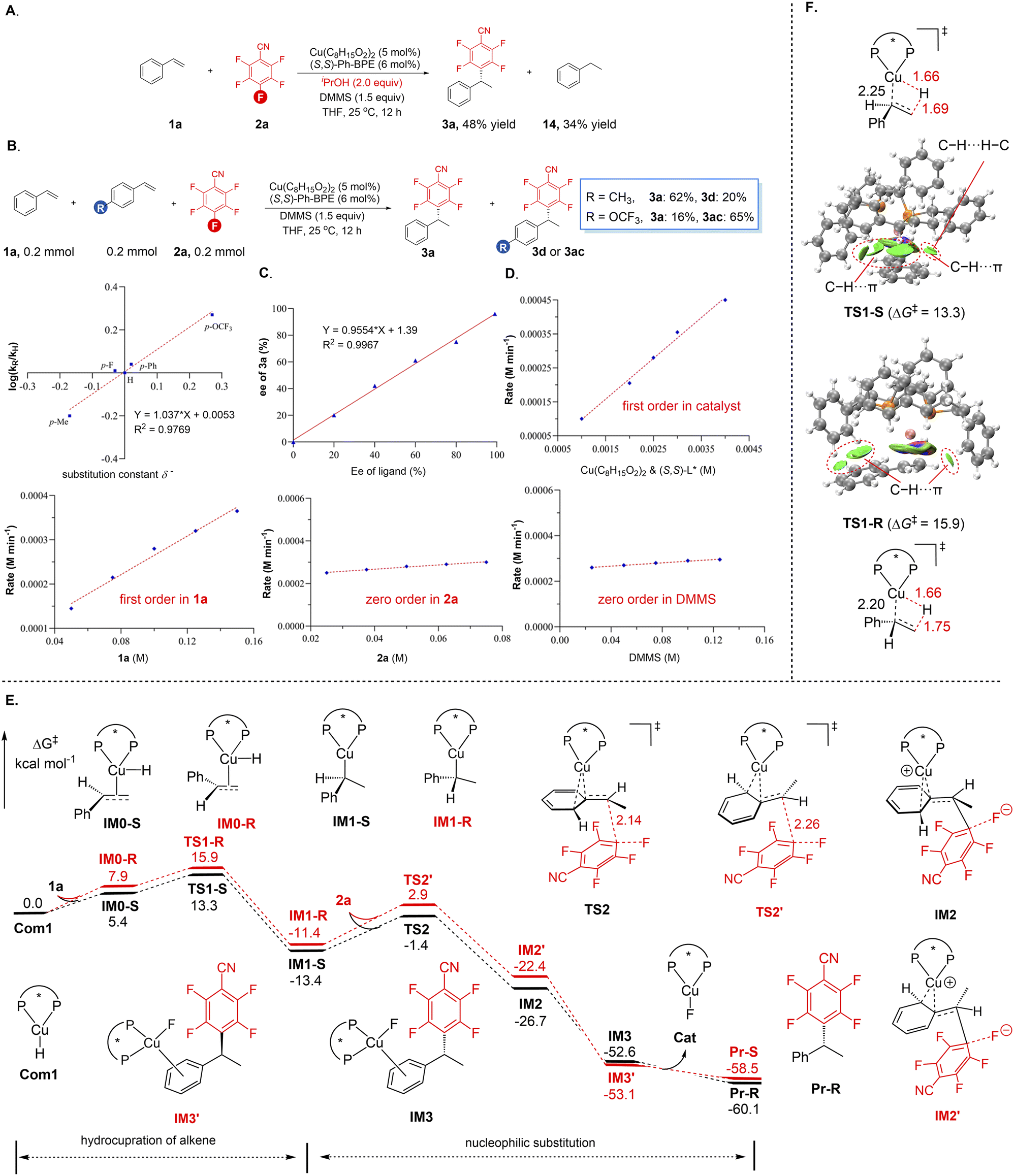 | ||
| Fig. 2 Mechanism investigation. | ||
This experimental observation indicates that the in situ-generated benzylic copper intermediate, derived from a regio- and enantioselective insertion of a chiral L*CuH species across the C–C double bond in the alkene, is involved in this transformation. Subsequently, comparative experiments were performed to further elucidate the properties of the possible intermediates (Fig. 2A). A competitive reaction system consisting of styrene and para-electron-donating or withdrawing functionality-substituted styrene derivatives with pentafluorobenzonitrile revealed that alkenes bearing an electron-withdrawing group on the aromatic ring exhibit higher reactivity, suggesting that the hydrometallation of the alkene serves as the rate-determining step in the catalytic cycle. Furthermore, a well-defined linear correlation between log(kR/kH) against substituent parameters (δ−) with a positive slope (ρ = 1.0) of the line was obtained, confirming the buildup and stabilization of negative charge in the transition state during the rate-determining step, consistent with the aforementioned intermolecular competitive experiment (Fig. 2B). Additionally, a nonlinear relationship experiment was investigated under the standard reaction conditions, supporting a monomeric copper complex bearing a single chiral ligand (Fig. 2C). Kinetic studies were then carried out to explore the dependency of the reaction rate on the concentrations of each reaction component, aiming to identify the turnover-limiting step of the reaction (Fig. 2D). The reaction exhibited first-order dependence on the concentrations of the catalyst and alkene, while displaying zeroth-order dependence on the concentrations of polyfluoroarenes and silane. These experimental data also suggested that the hydrometallation of the alkene serves as the rate-determining step, whereas the subsequent defluorinative alkylation process of polyfluoroarenes may not involve the rate-determining step. We next used DFT calculations to construct a mechanistic model to understand the reaction details and stereochemistry of this asymmetric defluorinative alkylation with styrene 1a and pentafluorobenzonitrile 2a as model reactions. The oxidation state of the copper catalyst in this catalytic system was initially determined. Control experiments manifested that both the Cu(I) salt and commercially available Cu(I)H catalyst, [CuH(PPh3)]6, were effective for this transformation (see Table S1). Taken together with previous reports,17,19,20 these results suggested that LCu(I)H is the most likely catalytic active species in this reaction. As depicted in Fig. 2E, a feasible catalytic cycle begins with the regio- and enantioselective insertion of the alkene into the well-established, in situ-generated (S,S)-Ph-BPE-coordinated CuH species (IM0-S and IM0-R). This process favorably affords the IM1-S intermediate via the transition state TS1-S (ΔG‡ = 13.3 kcal mol−1), which is 2.6 kcal mol−1 lower than that of the transition state TS1-R leading to the formation of the IM1-R intermediate. This theoretically calculated energy difference is in good agreement with the experimental result (96% ee). These intermediates, in accordance with previous demonstration,22 then undergo a sequence of 1,3-cupratropic shift and anti-SN2′-like process to attack the para-C–F bond in 2avia transition states TS2 (ΔG‡ = 12.0 kcal mol−1) and TS2′ (ΔG‡ = 14.3 kcal mol−1), delivering the intermediates IM2 and IM2′, respectively. The rapid transformation of the weakly associated fluoride ion with Cu(I) cation in a diffusion-limited, activationless process could yield IM3 and IM3′. Finally, the dissociation of the Cu(I) cation and fluoride ion from IM3 and IM3′ followed by their recombination, could release Cu(I)F and afford the corresponding Pr-R and Pr-S products. In accordance with the kinetic experiment observation, our calculations also identified that the hydrometallation of alkene serves as both the rate- and enantio-determining step making the Si-face more susceptible to the insertion with the chiral CuH species. Further analysis, at the M06/6-311+G(d,p)-SDD/SMD(THF)//B3LYP/D3(BJ)/6-31G(d)-SDD level of theory which could describe both the geometries and relative energies of the transition metal systems with reasonable accuracy, revealed that the two transition states TS1-R and TS1-S exhibit distinct relative energy primarily derived from the significant non-covalent interactions, such as C–H⋯π and C–H⋯H–C dispersion interactions (Fig. 2F). Independent gradient model analysis based on Hirshfeld partition (IGMH)23 reveals that the larger interaction energies in TS1-S (−6.7 kcal mol−1) are attributed to the stronger C–H⋯π interactions between the ligand's phenyl substituent and the substrate's –C–H and phenyl moieties. In contrast, these interactions are significantly weaker in TS1-R (−3.9 kcal mol−1), rationalizing the energetic preference for TS1-S.
Conclusions
In summary, we have successfully developed a copper-catalysed asymmetric defluorinative alkylation of polyfluoroarenes with alkenes, 1,3-dienes, and (Z/E)-mixed 1,3-dienes. This method demonstrates an excellent substrate scope, high functional group compatibility, and consistently outstanding regio- and stereoselectivity. This method provides a straightforward and mild route for constructing chiral Csp3–CArF bonds without the previous need for preformed organometallics. A series of experimental studies and DFT calculations have revealed the plausible mechanism and the origins of stereochemistry. We believe that this study will inspire further advancements in the synthesis of highly valuable yet challenging-to-access chiral polyfluoroarene compounds.Author contributions
T. Xiong and Q. Zhang designed the project and directed the work. D. Shi, Y. Jiang, S. Wang, J. Yin, X. Yuan, S. Ma, and X. Li performed all synthetic experiments, and L. Zhu performed all DFT calculations. T. Xiong, Q. Zhang, L. Zhu, J. Sun and D. Shi wrote the paper. All authors discussed, read and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Additional raw data are available from the corresponding author upon reasonable request. Supplementary information: all experimental procedures, computational details, and spectroscopic data. See DOI: https://doi.org/10.1039/d5sc08090h.Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (grant 2024YFA1509701), the National Natural Science Foundation of China (grants 22571037, 22171042, and 22193012), the Youth Talent Climbing Program of NENU and the Fundamental Research Funds for the Central Universities of Northeast Normal University.Notes and references
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- G. Balz and G. Schiemann, Ber. Dtsch. Chem. Ges. B, 1927, 60, 1186 CrossRef.
- G. C. Finger and C. W. Kruse, J. Am. Chem. Soc., 1956, 78, 6034 CrossRef CAS.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) Y. Li, G. S. Li and X. S. Wang, Adv. Synth. Catal., 2014, 356, 1412–1418 CrossRef CAS; (c) R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS.
- (a) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS; (b) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed; (c) M. Aizenberg and D. Milstein, Science, 1994, 265, 359–361 CrossRef CAS PubMed; (d) M. Aizenberg and D. Milstein, J. Am. Chem. Soc., 1995, 117, 8674–8675 CrossRef CAS; (e) T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius and A. Mix, J. Am. Chem. Soc., 2008, 130, 9304–9317 CrossRef CAS PubMed; (f) H. Lv, J. H. Zhan, Y. B. Cai, Y. Yu, B. Wang and J. L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216–16227 CrossRef CAS; (g) H. B. Lv, Y. B. Cai and J. L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3203–3207 CrossRef CAS PubMed; (h) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed; (i) J. Lu, N. S. Khetrapal, J. A. Johnson, X. C. Zeng and J. Zhang, J. Am. Chem. Soc., 2016, 138, 15805–15808 CrossRef CAS PubMed.
- (a) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed; (b) W. H. Guo, Q. Q. Min, J. W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9075–9078 CrossRef CAS; (c) X. W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470–12473 CrossRef CAS PubMed; (d) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313–14318 CrossRef CAS; (e) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed; (f) Y. M. Tian, X. N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS PubMed.
- (a) N. Yoshikai, H. Mashima and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 17978–17979 CrossRef CAS PubMed; (b) S. A. Macgregor, D. C. Roe, W. J. Marshall, K. M. Bloch, V. I. Bakhmutov and V. V. Grushin, J. Am. Chem. Soc., 2005, 127, 15304–15321 CrossRef CAS; (c) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS PubMed; (d) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505–19511 CrossRef CAS PubMed; (e) F. Zhu and Z. X. Wang, J. Org. Chem., 2014, 79, 4285–4292 CrossRef CAS PubMed; (f) D. Yu, L. Lu and Q. Shen, Org. Lett., 2013, 15, 940–943 CrossRef CAS PubMed; (g) M. Ohashi, R. Doi and S. Ogoshi, Chem.–Eur J., 2014, 20, 2040–2048 CrossRef CAS PubMed; (h) Z. J. Luo, H. Y. Zhao and X. Zhang, Org. Lett., 2018, 20, 2543–2546 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- V. V. Konovalov, S. S. Laev, I. V. Beregovaya, L. N. Shchegoleva, V. D. Shteingarts, Y. D. Tsvetkov and I. Bilkis, J. Phys. Chem. A, 2000, 104, 352–361 CrossRef CAS.
- (a) Y. Kiso, K. Tamao and M. Kumada, J. Organomet. Chem., 1973, 50, C12–C14 CrossRef CAS; (b) H. Guo, F. Kong, K. I. Kanno, J. He, K. Nakajima and T. Takahashi, Organometallics, 2006, 25, 2045–2048 CrossRef CAS; (c) T. Wang, B. J. Alfonso and J. A. Love, Org. Lett., 2007, 9, 5629–5631 CrossRef CAS PubMed; (d) A. D. Sun and J. A. Love, J. Fluorine Chem., 2010, 131, 1237–1240 CrossRef CAS; (e) Y. Sun, H. Sun, J. Jia, A. Du and X. Li, Organometallics, 2014, 33, 1079–1081 CrossRef CAS; (f) A. D. Sun, K. Leung, A. D. Restivo, N. A. LaBerge, H. Takasaki and J. A. Love, Chem.–Eur J., 2014, 20, 3162–3168 CrossRef CAS PubMed; (g) S. H. Xiao, Y. Xiong, X. X. Zhang and S. Cao, Tetrahedron, 2014, 70, 4405–4411 CrossRef CAS; (h) D. Yu, C. S. Wang, C. Yao, Q. Shen and L. Lu, Org. Lett., 2014, 16, 5544–5547 CrossRef CAS PubMed; (i) T. Iwasaki, A. Fukuoka, X. Min, W. Yokoyama, H. Kuniyasu and N. Kambe, Org. Lett., 2016, 18, 4868–4871 CrossRef CAS PubMed; (j) T. Iwasaki, K. Okamoto, H. Kuniyasu and N. Kambe, Chem. Lett., 2017, 46, 1504–1507 CrossRef CAS; (k) T. Iwasaki, X. Min, A. Fukuoka, L. Zhu, R. Qiu, T. Yang, M. Ehara, A. Sudalai and N. Kambe, J. Org. Chem., 2018, 83, 9267–9277 CrossRef CAS PubMed.
- (a) A. Singh, J. J. Kubik and J. D. Weaver, Chem. Sci., 2015, 6, 7206–7212 RSC; (b) S. Priya and J. D. Wwaver III, J. Am. Chem. Soc., 2018, 140, 16020–16025 CrossRef CAS PubMed; (c) A. Arora and J. D. Weaver, Acc. Chem. Res., 2016, 49, 2273–2283 CrossRef CAS PubMed.
- J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashimi, Angew. Chem., Int. Ed., 2017, 56, 7266–7270 CrossRef CAS PubMed.
- (a) X. Sun and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 10557–10562 CrossRef CAS; (b) W. Xu, Q. Shao, C. Xia, Q. Zhang, Y. Xu, Y. Liu and M. Wu, Chem. Sci., 2023, 14, 916–922 RSC; (c) H. Zhang, Q. Shao, Q. Zhang, C. Xia, W. Xu and M. Wu, Org. Lett., 2023, 25, 4556–4561 CrossRef CAS PubMed; (d) M. Wang, D. Pan, Q. Zhang, Y. Lei, C. Wang, H. Jia, L. Mou, X. Miao, X. Ren and Z. Xu, J. Am. Chem. Soc., 2024, 146, 6675–6685 CrossRef CAS.
- X. Yi, R. Mao, L. Lavrencic and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 23557–23563 CrossRef CAS.
- F.-P. Wu, X.-W. Gu, H.-Q. Geng and X.-F. Wu, Chem. Sci., 2023, 14, 2342–2347 RSC.
- (a) Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS PubMed; (b) P. Cheruku, A. Paptchikhine, T. L. Church and P. G. Andersson, J. Am. Chem. Soc., 2009, 131, 8285–8289 CrossRef CAS PubMed; (c) J. Yang, L. Massaro, S. Krajangsri, T. Singh, H. Su, E. Silvi, S. Ponra, L. Eriksson, M. S. G. Ahlquist and P. G. Andersson, J. Am. Chem. Soc., 2021, 143, 21594–21603 CrossRef CAS PubMed; (d) J. Yang, L. Massaro, W. Hu, B. C. P. Peters, N. Birke, C. Chantana, T. Singh and P. G. Andersson, J. Am. Chem. Soc., 2023, 145, 626–633 CrossRef CAS PubMed; (e) R. Mao, D. M. Taylor, D. J. Wackelin, T. Rogge, S. J. Wu, K. M. Sicinski, K. N. Houk and F. H. Arnold, Nat. Synth., 2024, 3, 256–264 CrossRef CAS PubMed; (f) P. Lu, H. Wang, Y. Mao, X. Hong and Z. Lu, J. Am. Chem. Soc., 2022, 144, 17359–17364 CrossRef CAS PubMed; (g) Y. Bao, C. Zheng, K. Xiong, C. Hu, P. Lu, Y. Wang and Z. Lu, J. Am. Chem. Soc., 2024, 146, 21089–21098 CrossRef CAS PubMed; (h) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746–6749 CrossRef CAS PubMed; (i) K. X. Zhang, M. Y. Liu, B. Y. Yao, Q. L. Zhou and L. J. Xiao, J. Am. Chem. Soc., 2024, 146, 22157–22165 CrossRef CAS PubMed.
- (a) J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS; (b) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (c) M. T. Pirnot, Y. M. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 48–57 CrossRef CAS PubMed; (d) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed; (e) R. Y. Liu and S. L. Buchwald, Acc. Chem. Res., 2020, 53, 1229–1243 CrossRef CAS PubMed.
- (a) S. D. Früs, M. T. Pirnot, L. N. Dupuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 7242–7246 CrossRef PubMed; (b) S. Yu, H. Leng and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 15896–15900 CrossRef CAS PubMed; (c) M. W. Gribble, J. S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057–5060 CrossRef CAS PubMed; (d) M. Seo, S. Seo, J. Jung and H. Kim, Angew. Chem., Int. Ed., 2025, 64, e202420918 CrossRef CAS PubMed.
- X. Li, B. Fu, Q. Zhang, X. Yuan, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23056–23060 CrossRef CAS PubMed.
- (a) B. Fu, Y. Zhao, X. Yuan, Y. Li, J. Yin, S. Wang, T. Xiong and Q. Zhang, Chin. Chem. Lett., 2024, 35, 108372 CrossRef CAS; (b) B. Fu, L. Wang, K. Chen, X. Yuan, J. Yin, S. Wang, D. Shi, B. Zhu, W. Guan, Q. Zhang and T. Xiong, Angew. Chem., Int. Ed., 2024, 63, e202407391 CAS; (c) Y. Wang, J. Yin, Y. Li, X. Yuan, T. Xiong and Q. Zhang, ACS Catal., 2022, 12, 9611–9620 CrossRef CAS; (d) S. Wang, K. Chen, J. Niu, X. Guo, X. Yuan, J. Yin, B. Zhu, D. Shi, W. Guan, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202410833 CAS.
- J. Yin, X. Yuan, S. Wang, D. Shi, S. Ma, L. Zhu, X. Li, Q. Zhang and T. Xiong, J. Am. Chem. Soc., 2025, 147, 25592–25602 CrossRef CAS PubMed.
- X. Zhao, W. Y. Tong and X. Wang, Org. Chem. Front., 2023, 10, 5651–5659 RSC.
- (a) T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed; (b) Y. Li, D. Liu, X. Hu, J. Y. Zhang, Q. W. Zhu, B. Men, G. W. Gao, P. W. Chen, Y. Z. Tong, Z. Chang, Z. Li, X. Lu and Y. Fu, Nat. Synth., 2024, 3, 1134–1144 CrossRef CAS; (c) X. Hu, C. Wang, L. Yu, Y.-Z. Tong, Z. Li, Y. Li, Z. Chang, J.-Y. Zhang, J. Kuang, D. Liu, C. Tian, Y.-H. Xu, X. Lu and Y. Fu, Nat. Synth., 2025, 4, 1442–1452 CrossRef CAS; (d) M. Zhao, L. Zhu, Q. Zhang and J. Zhang, Org. Chem. Front., 2025, 12, 4986–4996 RSC; (e) B. Liu, D. Liu, X. Rong, X. Lu, Y. Fu and Q. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218544 CrossRef CAS; (f) M. Shen, C. Niu, X. Wang, J.-B. Huang, Z. Zhao, S.-F. Ni and Z.-Q. Rong, JACS Au, 2024, 4, 2312–2322 CrossRef CAS PubMed; (g) M. Zhao, H. Yuan and J. Zhang, Inorg. Chem., 2024, 63, 21031–21041 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |