
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling optoelectronic properties and aggregation of planar dipoles through symmetry-preserving modifications
José
García-Calvo
 *ab,
María
Modino-Montes
a,
Ignacio
Romero-Muñiz
a and
Tomás
Torres
*abc
*ab,
María
Modino-Montes
a,
Ignacio
Romero-Muñiz
a and
Tomás
Torres
*abc
aDepartment of Organic Chemistry, Facultad de Ciencias, Universidad de Madrid Autónoma Cantoblanco, 28049 Madrid, Spain. E-mail: jose.garciac@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Campus de Cantoblanco, 28049 Madrid, Spain
cIMDEA-Nanociencia, c/Faraday 9, Campus de Cantoblanco, 28049 Madrid, Spain
First published on 14th November 2025
Abstract
Planar and symmetric molecular architectures with defined donor–acceptor distributions offer exceptional versatility for optoelectronic applications. Perylene monoimides exemplify these features, and here we systematically explore strategies to tune their dipole-like distribution while preserving symmetry. Correlating optical properties in solution and solid states with electron-donating and/or -withdrawing groups at the ortho positions reveals how subtle modifications control the intermolecular interactions and therefore, aggregation and optoelectronics. Single-crystal X-ray diffraction further uncovers distinct stacking modes, highlighting the decisive role of molecular design in tailoring functionality for diverse applications.
Introduction
When developing new optoelectronic systems from polycyclic aromatic hydrocarbons (PAHs), characteristics such as their planarity, symmetry or charge distribution are crucial for their performance. Perylene derivatives (PDs) are a paradigmatic example1–3 and, as such, their properties have been extensively investigated.3 Additionally, PDs show exceptional photothermal and chemical stability, photoluminescent absorption and emission in the UV-vis-IR region as well as a pronounced tendency to self-assemble.4–6 Focusing on the differences between the most common derivatives, bare perylene is a known electron-rich molecule that tends to act as a donor, while perylene diimides (PDIs) are electron poor, and therefore acceptors. This feature in particular provides complementary applications in a wide range of fields,7 spanning from organic materials8,9 to cellular biology.10 With that in mind, the mixed derivatives, perylene monoimides (PMIs), are particularly attractive, since they possess a unique dipole-like charge distribution and benefit from well-established synthetic methodologies,11 similar to perylene or PDIs.12 PMIs also exhibit mixed reactivity that facilitates selective derivatization and unique features regarding electronic distribution and redox potentials.12 These attributes have led to their use in the design of organic photovoltaic cells (OPVs) and organic field-effect transistors (OFETs).13,14 Therefore, and besides the usual pery- and bay- substitutions,15 modifying the donor–acceptor properties in PMIs with a symmetry-preserving alternative represents an unexplored and very appealing objective to understand the potential of this molecular design. To accomplish that, a systematic procedure for the selective borylation of PMIs at the ortho positions has been recently optimized, in which two scenarios for ortho-modification coexist.16 On one hand, derivatization may be performed at positions 2 and 5 (Scheme 1), which are ortho- to the imide group, resulting in direct intramolecular interactions with it or its N-substituents. On the other hand, ortho-substitution at positions 8 and 11 is barely explored in the literature,17 while being in the donor part of the molecule and conferring an increased degree of rotational freedom around the attachment point. | ||
Scheme 1 General synthetic procedure for compounds 2a, 2b, 3a, 3b, 4a and 4b. (a) B2Pin2 (8 eq.), [Ir(OMe)(COD)]2 (0.05 eq.) and P(C6F5)3 (0.1 eq.), dioxane (20 mM), 140 °C 72 h; (b) B2Pin2 (8 eq.), [Ir(OMe)(COD)]2 (0.05 eq.) and dtbpy (0.1 eq.), THF (20 mM), 110 °C (MW), 1 h. (c) Aryl-halide (4 eq.), K2CO3 (10 eq.), Pd2(dba)3 (0.2 eq.), SPhos (0.4 eq.), toluene/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 36 h. :1), 36 h. | ||
Previous research into ortho-substituted perylene derivatives also provides valuable insights into their optoelectronic properties. For instance, Marder and collaborators reported the influence of incorporating tertiary amines at the ortho-positions of a perylene core.18 Similarly, Wasielewski and collaborators described a terrylenediimide19 and a PDI20 featuring ortho-substituted phenyl groups. Specifically concerning PMIs, aliphatic chains at positions 2 and 5 have been used to control solubility/aggregation21 and also a bis-phenyl substituted derivative at positions 2 and 5 has been explored for singlet fission applications.22 On the other hand, the introduction of donor groups, such as –OMe, or acceptor groups, such as –CF3, has demonstrated to be an optimal approach to regulate the brightness or fluorescence quantum yield in solution23 while it certainly plays a role in the solid properties too.
Consequently, studying the synthetic scope of these derivatives, as well as performing absorbance and photoluminescence studies, has proven to be an essential starting point for the development of optoelectronic materials with enhanced applications.24
Synthesis and characterization
PMI 1 and ortho-borylated compounds 5a and 5b (Scheme 1) were synthesized following previously reported procedures.16 First, 1 was produced from perylene dianhydride (PDA) by the method described in ref. 11. After that, based on a recent publication from our group,16 the selective ortho-borylation of PMI derivatives was performed (Section 2 of the SI). Ortho-borylation on the imide side (positions 2 and 5) was performed by refluxing at 140 °C a mixture of B2(Pin)2 (8 eq.), [Ir(Ome)(COD)]2 (0.4 eq.) and P(C6F5)3 (0.8 eq.) in dioxane for 72 hours, to obtain 5a (72%). To attain the opposite ortho-diborylation, 5b (85%, positions 8 and 11), the same reagents were used, but the ligand was exchanged with 4,4-di-tert-butyl-2,2-dipyridyl (dtbpy, 0.4 eq.), THF was used as the solvent and the reaction was heated (110 °C) in the MW for one hour.Compounds 6a and 6b were obtained under the same reaction conditions as 5a, however starting from 3a and 3b, and leading to similar yields (79% and 77%, respectively). Improvements from previously reported procedures were achieved by selective precipitation of the borylated PMIs. The reaction crudes of 5a, 5b, 6a and 6b were dissolved in mixtures CH2Cl2/MeOH (10:1) and, after the evaporation of CH2Cl2, the pure products were isolated by filtration under vacuum. Next, Suzuki couplings with different halide-phenyl rings were performed following standard conditions. Into a flask containing borylated PMI; K2CO3 (10 eq.), SPhos (0.4 eq.), Pd2(dba)3 (0.4 eq.) and the corresponding halide, 1-bromo-3,5-dimethoxy-benzene or 1-bromo-3,5-ditrifluoromethyl-benzene (0.4 eq.), were added. After 36 h stirring at 110 °C in a toluene/water (4:1) mixture, the crude was diluted with CH2Cl2, washed with water, filtered, concentrated under vacuum and purified by silica gel column chromatography (CH2Cl2/heptane → CH2Cl2) to obtain the products in good to excellent yields, 2a (61%), 2b (70%), 3a (51%), 3b (65%), 4a (79%) and 4b (77%).
Results and discussion
X-ray crystal diffraction structures
Here, the X-ray diffraction results of single crystal structures are reported for 2a, 4a and 4b. Their molecular structures were confirmed and the intermolecular interactions were studied. The crystals were obtained by methanol diffusion to a saturated sample in CH2Cl2 and measured under the conditions detailed in the SI (Section 7).Compound 4a was obtained as a triclinic system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while 2a and 4b were monoclinic and P21/c. In compound 4a, the crystal showed a slip-stacked pattern (Fig. 1a and b) to accommodate the steric hindrance caused by the substituents. Adjacent molecules interact via π–π stacking, with a π–π distance of 3.4 Å and a slippage of 3.3 Å and 1.3 Å, along the long and short axis, respectively. In contrast, 4b displayed discrete dimeric structures, with both perylene cores aligned with opposite orientations but with longer distances between them, 3.6 Å (Fig. 2d and e). Compound 2a presented a stacking structure more closely resembling 4a (See Section 7.1 of the SI). However, in 2a (Fig. S60), the substituents were accommodated with a 29° rotation from the imide position and showed higher but regular core–core distances (3.5 Å).
, while 2a and 4b were monoclinic and P21/c. In compound 4a, the crystal showed a slip-stacked pattern (Fig. 1a and b) to accommodate the steric hindrance caused by the substituents. Adjacent molecules interact via π–π stacking, with a π–π distance of 3.4 Å and a slippage of 3.3 Å and 1.3 Å, along the long and short axis, respectively. In contrast, 4b displayed discrete dimeric structures, with both perylene cores aligned with opposite orientations but with longer distances between them, 3.6 Å (Fig. 2d and e). Compound 2a presented a stacking structure more closely resembling 4a (See Section 7.1 of the SI). However, in 2a (Fig. S60), the substituents were accommodated with a 29° rotation from the imide position and showed higher but regular core–core distances (3.5 Å).
 | ||
| Fig. 1 Single crystal X-ray structure of compound 4a (a–c) and 4b (d–f) showing the shortest intermolecular packing of two stacked molecules from the perpendicular (a and d) or parallel (b and e) point of view. | ||
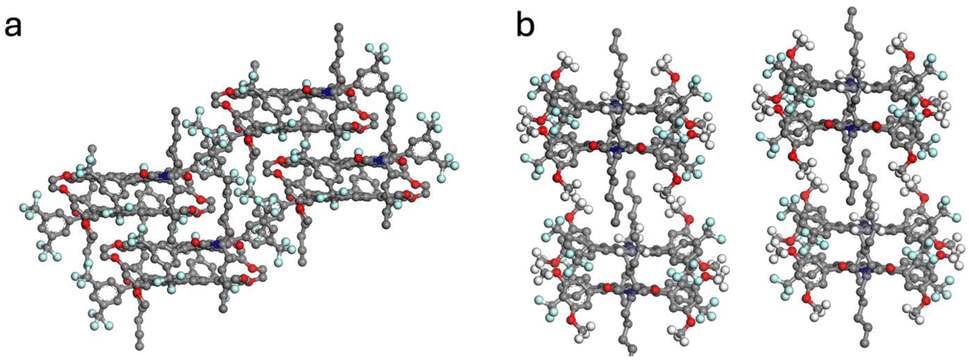 | ||
| Fig. 2 X-ray diffraction packing of 4a (a) and 4b (b), perpendicular view to the stack direction. | ||
Analysis of the crystalline structure also provides insights to understand the optical properties (vide infra). Compound 4a (Fig. 2a) revealed a good match but inverted distribution with an ortho-phenyl-substituted PMI reported by the group of Wasielewski.22 Specifically, the distances and angles between adjacent perylene cores were comparable. The results were slightly different for compound 2a. A similar stacking pattern was observed (Fig. S61) but with different distances and torsional arrangements, more similar to other perylene derivatives25 and in line with a π-stack aggregate.26 These results agree with the polymorphism observed in less substituted PDIs,27 since changes in the structure determine the supramolecular interactions and therefore the crystalline patterns. Conversely, the crystalline structure of 4b (Fig. 2b) showed greater similarities to PMI derivatives whose aggregation behavior is primarily governed by extended chains at the peri-positions28 or with PDI cages/dimers, with structures mostly determined by the covalent bonds connecting two PDI moieties.26,29,30
Photophysical properties
The presence of ortho-substituents in PDs is known to exert a strong influence over their optoelectronic properties, requiring a differentiated study between solid22 and dissolved samples.31 Starting with the dissolved samples (Fig. 3), the absorbance spectra of PMI derivatives with different donor (D) and acceptor (A) topologies were studied. 2a (AA–D), 2b (DA–D), 3a (A–DD), 3b (A–DA), 4a (AA–DD) and 4b (DA–DA) showed similar spectra to the unsubstituted PMI 1 (A–D) (Fig. 4a–c and Section 5.1 of the SI). They had a S0 → S1 transition attributed to the HOMO → LUMO, with no significant contribution from other processes, as confirmed in TD-DFT calculations (Section 5.1 of the SI) and showed extinction coefficients (ε) between 27 and 40 mM−1 cm−1 (Table 1). Additionally, similar to many perylene or PDI derivatives,32,33 a π → π* transition was observed with the usual four distinct peaks, associated to the vibronic progression of the 0 → 1 transition.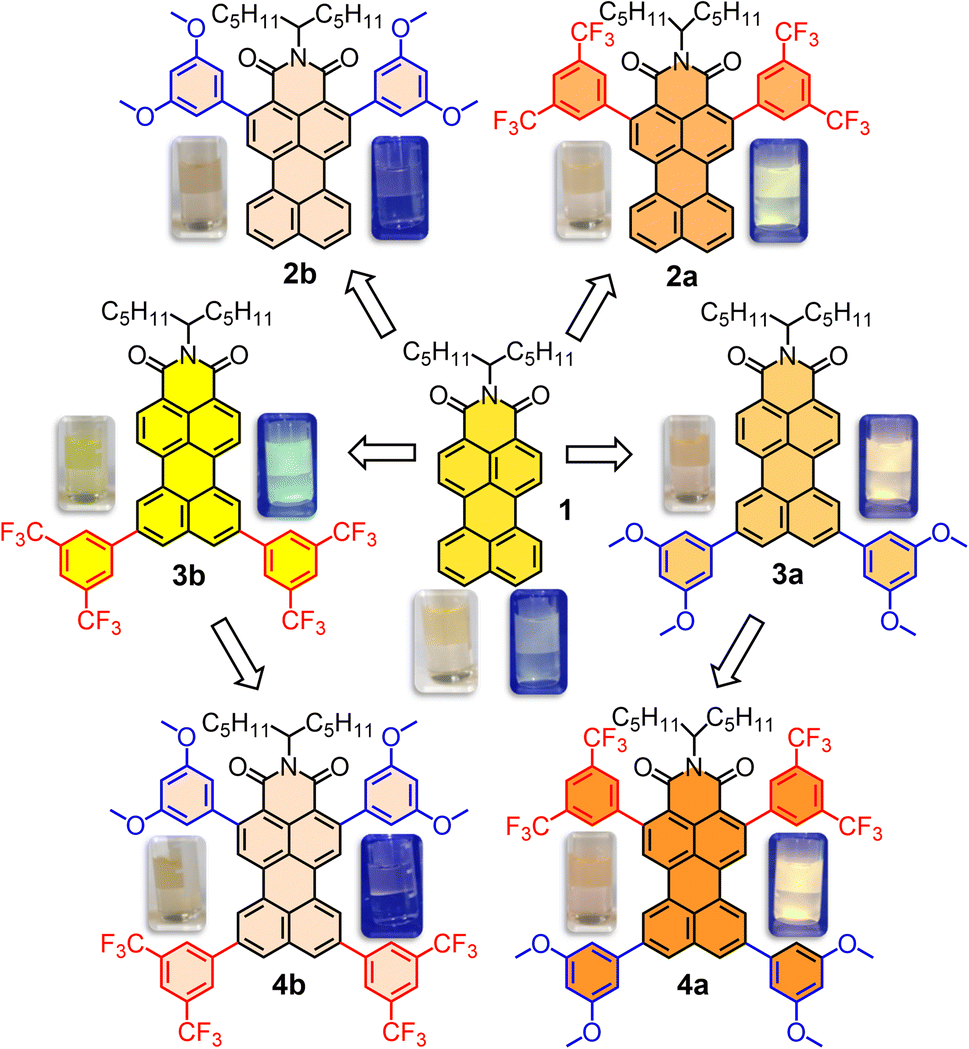 | ||
| Fig. 3 Structure and picture of dissolved samples (10 µM) of compounds 1, 2a, 2b, 3a, 3b, 4a and 4b; under visible light and under UV light (366 nm lamp). | ||
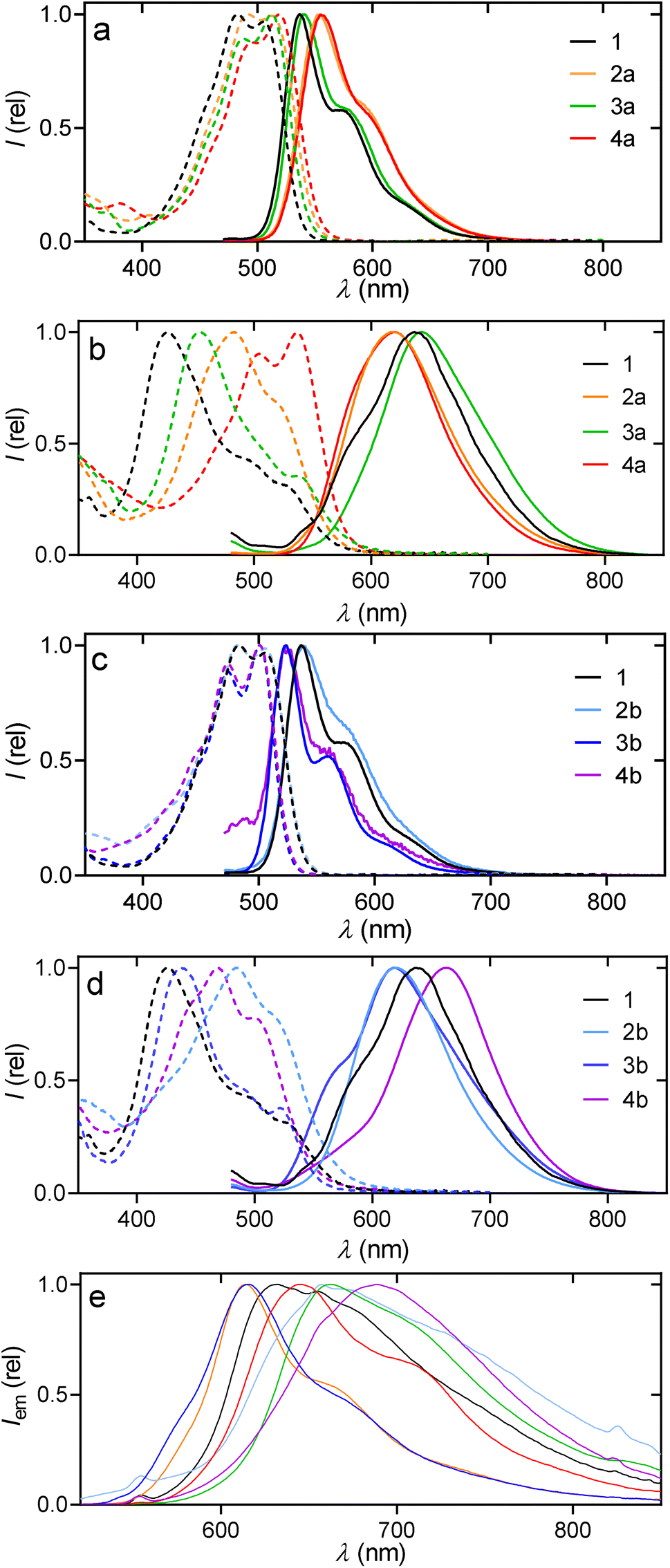 | ||
| Fig. 4 Normalized absorption (dashed) and emission intensity (plain) in chloroform ((a and c) 5 µM, λex = 460 nm), in THF/water (1:99, (b and d) 10 µM, λex = 460 nm) and in the solid state ((e) λex = 450 nm) of compounds 1 (black), 2a (orange), 2b (light blue), 3a (green), 3b (dark blue), 4a (red) and 4b (purple). | ||
| Cpda | ε | λ ab | λ em | Δλabe | Δλemf | Φ d | FWHMh | τ F | k r | λ ab | λ em | λ em | Φ s | Topologyo |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Compound.
b Molar extinction coefficients (mM−1 cm−1) in chloroform, at the maximum of absorption.
c Wavelength (nm) of the maximum of absorption in chloroform.
d Wavelength (nm) of the maximum of emission (λex = 460 nm) in chloroform.
e Variation (nm) of the maximum of absorption between DMSO vs. MCH.
f Variation (nm) between the maximum of emission in DMSO vs. MCH.
g Photoluminescence quantum yield (%) in chloroform.
h Full width at half maximum of absorption in chloroform.
i Fluorescence lifetime in chloroform.
j Radiative (fluorescence) rate constant.
k Wavelength (nm) of the maximum of absorption in THF/water (1:99).
l Wavelength (nm) of the maximum of emission (λex = 460 nm) THF/water (1:99).
m Wavelength (nm) of the maximum of emission in the solid state (λex = 460 nm).
n Photoluminescence quantum yield (%) in the solid state.
o Donor (D) and acceptor (A).
|
||||||||||||||
| 1 | 39 | 483 | 537 | 6 | 49 | 79 | 75.5 | 5.06 | 0.156 | 426 | 637 | 631 | 6 | A–D |
| 2a | 31 | 492 | 554 | 7 | 50 | 78 | 78 | 5.56 | 0.140 | 483 | 619 | 614 | 13 | AA–D |
| 2b | 28 | 483 | 538 | 10 | 49 | 4 | 78 | 4.37 | 0.009 | 485 | 620 | 658 | 1 | DA–D |
| 3a | 38 | 512 | 539 | 10 | 45 | 78 | 69 | 4.91 | 0.159 | 454 | 643 | 661 | 5 | A–DD |
| 3b | 40 | 501 | 524 | 16 | 49 | 86 | 80 | 4.60 | 0.187 | 438 | 619 | 615 | 6 | A–DA |
| 4a | 34 | 519 | 555 | 7 | 44 | 68 | 70 | 5.39 | 0.126 | 537 | 620 | 644 | 10 | AA–DD |
| 4b | 34 | 501 | 522 | 17 | 39 | 0.4 | 72.5 | 4.17 | 0.001 | 469 | 663 | 690 | 3 | DA–DA |
In contrast to the analogous ε, the shape of the absorption spectra and the wavelengths of both the absorption and fluorescence emission were highly affected by the substituents and their positions (Fig. 3 and 4a–c). First, two clearly different vibronic distributions were observed, on one hand 1, 2a and 2b have their maxima of absorption located at the second less energetic peak, while all the compounds with substituents in positions 8 and 11 had their maxima at the less energetic one (Table 1). Additionally, when looking for a bathochromic change in absorption, the introduction of 3,5-trifluoromethyl-phenyl groups in positions 2 and 5 (2a) resulted in a more effective methodology than when the donor 3,5-dimethoxy-phenyl group was at positions 8 and 11 (2b), +8 nm vs. +7 nm (Table 1). In contrast, introducing the same acceptor and donor in the reversed positions produced a hypsochromic shift for compound 3b (−4 nm) while no shift was observed for 2b. Finally, the presence of tetra-substituted compounds produced exactly the sum of the individual contributions in both 4a (+14 nm) and 4b (−4 nm).
All derivatives, when dissolved, emitted fluorescence with the maximum in the region of 500–600 nm. The shifts in emission followed the same trends as absorptions, with a strong response to changes in donor–acceptor properties. Red-shifted emissions for 2a (+17 nm), 3a (+2 nm) and 4a (+18 nm) were observed, while they were blue-shifted for 3b (−13 nm) and 4b (−15 nm). The Stokes shift from PMI 1 (32 nm) was also maximized for compounds 2a (41 nm) and 4a (36 nm), while it decreased for 3a (27 nm), 3b (23 nm) and 4b (21 nm).
The photoluminescence quantum yields (ΦF) were measured in chloroform and compared to PMI 1 (79%). Compounds 2a (78%) and 3a (78%) did not show significant changes in ΦF. On the other hand, compound 3b, with phenyl–CF3 acceptors at positions 8 and 11, displayed an increase in emission to 86%, contrary to the expected decrease due to the rotational freedom of the substituents, while this prediction was only true for tetrasubstituted 4a, with its ΦF slightly reduced (68%). However, the most impactful changes were observed for 2b (4%) and even more for 4b (0.4%), where almost complete inhibition of fluorescence was caused by the 3,5-dimethoxy-phenyl substituents ortho to the imide group. This kind of inhibition was, again, not predicted by experimental methods such as the increase of the full width at half maximum (FWHM, Table 1) which it is taken as an indicator of a decrease in ΦF.23 However, in the literature, a decrease has been observed for perylene and PDIs when introducing donor groups at ortho positions,31,33 although it was less significant. For compounds 2b and 4b, we attribute the decrease to a non-radiative relaxation process, involving the close proximity of the HOMO and HOMO−1 orbitals (vide infra), maximized in 4b, which produced alternative relaxation pathways without fluorescent emission.
Fluorescence lifetimes (τF) were also measured for all the compounds (Section 5.2 of the SI), evidencing a decrease in the fluorescence time from 2a (5.6 ns) > 4a (5.4 ns) > 1 (5.1 ns) > 3a (4.9 ns) > 3b (4.6 ns) > 2b (4.4 ns) > 4b (4.2 ns). Rate-constants (kr) were between 1.2 and 2 × 108 s−1, for all compounds but the barely emissive 2b and 4b (see Table 1). These kr values were akin to other highly emissive perylene or PDI derivatives. In conclusion, to maximize a radiative decay in solution the substitution at positions 8 and 11 was the best approach, while the derivatives substituted at positions 2 and 5 increased the non-radiative decay in all cases.
All derivatives presented a marked solvatochromic effect (Table 1 and Section 5.1 of the SI) between polar solvents (dimethyl sulfoxide, DMSO) and apolar ones (methylcyclohexane, MCH), with shifts between 6 and 17 nm in absorption and 39 to 50 nm in emissions. Among these, compound 4b was the one with the most noticeable change in absorption (17 nm) but the smallest in emission (39 nm) (Table 1, and Fig. S27).
In addition to the dissolved samples, solid state experiments of emission, excitation and photoluminescence quantum yields were also performed. Saturated solutions in dichloromethane were subjected to methanol diffusion, after which the samples were filtered to obtain the solids employed in the measurements. As observed in Fig. 4e, the spectra of PMIs were always in the 600–800 nm region and red-shifted an average of 100 nm compared to their performance when dissolved. All the spectra were broader, while the fine structure could still be well distinguished for 2a, 3b and 4a. Aiming to relate dissolved and solid-state studies, aggregation experiments in solvent mixtures were also performed. Since the solubility in organic solvents is moderate/high at c < 10−4 M, mixtures CHCl3/MeOH and CHCl3/MCH with up to 99% of MeOH and MCH, respectively, were studied. However, those mixtures did not show significant changes in shape for neither the absorbance nor the fluorescence (see SI, Section S5.3). In contrast, THF/H2O mixtures (10−5 M, Fig. 4b and d and detailed in Section S5.3) showed systematic changes, corresponding to aggregation whenever the solvent mixtures contained over 40% of water.
When all compounds were aggregated in solution (water/THF 99:1) or in solid-state, their emission spectra became generally broader and less defined compared to their dissolved forms, though the compounds retained consistent tendencies regarding spectral changes (Fig. 4). Specifically, samples 3a (643 nm/661 nm) and 4b (663 nm/690 nm) exhibited the most red-shifted emission maxima relative to the dissolved samples, whether aggregated or in the solid state. Conversely, 2a (619 nm/614 nm) and 3b (619 nm/615 nm) showed a slight blue-shift when comparing their aggregates to the solid-state emission; notably, these two compounds, along with 4a, also displayed a more defined fine-structure in their solid-state emission. Significantly, the aggregate emission spectrum of sample 3b was almost a perfect match with the solid-state emission spectrum in both shape and fine-structure (Fig. 4d and e). It is also worth noticing that photoluminescence quantum yields in solid samples (Φs) of perylene derivatives, especially in PDIs, are usually quenched due to the H-aggregates that are formed by π–π stacking, what explains the lower and broader emissions obtained.34 The Φs were very similar for pure PMI 1 (6%), 3a (5%) and 3b (6%) which are common values for fluorescent organic molecules in the solid state. The lowest values were, yet again, 2b (1%) and 4b (3%). In contrast, samples 2a and 4a reached 13% and 10%, respectively. Complementary, the absorption experiments of the aggregates in THF:water mixtures (Section 5.3 of the SI) show a tendency for aggregation more pronounced in compounds 1, 3a and 3b (formed at lower water content), which is explained by the more planar configuration that would facilitate aggregation in a similar manner. This is also supported by their analogous blue-shifted absorption spectra, corresponding to aggregated structures that may vary from dimers to π–π stacked oligomers, matching with the observed changed in the emission spectra (solution-aggregate-solid) and also with the results from the X-ray diffraction analysis as well as in the literature for PDI scaffolds.26 Considering that the three-dimensional arrangement of the molecules is key to their performance, these results highlight the substitution close to the imide group as the most reliable approach to achieving a higher efficiency in emissive PMI solid samples while going for or against the dipole does not necessarily provide a red or blue shift, respectively.
Electrochemical studies
Cyclic voltammetry studies were conducted with CH2Cl2, NBu4(PF6) (0.1 M), revealing two reduction potentials and up to two oxidation potentials for each of the PMI (Fig. S9–S12) with respect to the Fc/Fc+ standard. The oxidation and reduction potentials were taken from the differential pulse voltammetry (DPV) (Table 2) and, when compared with the values calculated from DFT, were transformed according to the literature.35,36| Ca | E h [o]1b | E h [o]2c | E h [r]1d | ΔEhe | H | L | µ |
|---|---|---|---|---|---|---|---|
| a Compound. b First halfway oxidation potential (V). c Second halfway oxidation potential (V). d First halfway reduction potential (V). e HOMO–LUMO gap calculated from the difference of reduction and oxidation potentials (V). f HOMO energy (eV) calculated by DFT (eV). g LUMO energy (eV) calculated by DFT. h Calculated dipolar moment. | |||||||
| 1 | 0.75 | — | −1.64 | 2.39 | −5.49 | −2.80 | 8.6 |
| 2a | 1.00 | — | −1.33 | 2.33 | −5.68 | −3.02 | 9.6 |
| 2b | 0.90 | 1.22 | −1.46 | 2.36 | −5.39 | −2.65 | 4.7 |
| 3a | 0.85 | 1.03 | −1.42 | 2.27 | −5.46 | −2.81 | 8.7 |
| 3b | 1.05 | — | −1.35 | 2.40 | −5.69 | −2.99 | 3.3 |
| 4a | 0.86 | 1.06 | −1.28 | 2.13 | −5.63 | −3.03 | 10.8 |
| 4b | 1.04 | 1.14 | −1.35 | 2.39 | −5.70 | −3.00 | 1.7 |
The first oxidation potential was, in all cases, anodically shifted from 0.75 V in PMI 1 to 0.85 V (3a and 4a), 0.90 V (2b), 1.00 V (2a), 1.04 V (4b) and 1.05 V (3b), showing that the presence of the acceptor, 3,5-trifluoromethyl-phenyl group, produced a more significant shift on the potential. Notably, a second halfway oxidation potential was observed for 3a (1.03 V), 4a (1.06 V) and 4b (1.14 V) with small gaps of 0.18 V, 0.20 V and 0.10 V, respectively. These results were in line with the computational estimations, with almost overlapping HOMO and HOMO−1 levels (vide infra), which made them almost degenerate and were also related to the observed optical properties.
As a counterpart, two peaks of reduction were detected under the conditions of the experiments (Table 2, and Section 4 for the SI), which typically match the results of PDIs. The first reduction potential shifted to less negative values when preserving the polarity of PMI 1, from −1.64 V in 1 to −1.42 V in 3a, to −1.33 V in 2a and to −1.28 V in 4a. In this case, the gap to the second reduction potential remained wide in all cases, between 0.39 V (4b) and 0.48 V (2a).
Finally, the calculated redox gap corresponding to a HOMO–LUMO transition was consistent with literature values for PDs, typically ranging from 2.4 to 2.2 V.33,37 Our findings indicated that these gaps remained largely invariant for compounds 1 (2.39 V), 2b (2.36 V), 3b (2.40 V), and 4b (2.39 V). Conversely, a notable decrease was observed for compounds 2a (2.33 V) > 3a (2.27 V) > 4a (2.13 V), thereby demonstrating the differential impact of modifications, depending on their alignment with the intrinsic polarity of the PMI core.
Computational calculations
Density functional theory (DFT) calculations were performed (B3LYP) at the 6-31G(d,p) level, with chloroform as the solvent. This basis set was selected since it has proven to perform well predicting the results of small changes in the structure, but without requiring highly demanding optimization methods. Additionally, it allows comparison with previous publications about perylene or PMI derivatives.16,38–40 The calculated HOMO–LUMO energy gap (Δ(H–L)) for the PMIs demonstrated consistency with experimental results and exhibited values analogous to those reported for ortho substituted PDI derivatives.41 First, a reduction in the band gap was systematically obtained 4a > 3a > 2a > 1, going from 2.60 to 2.69 eV (Fig. 5 and S8) and matching the reduction in the dipolar moment. Second, the modifications designed to oppose the PMI dipole moment succeeded in that regard 1 > 2b > 3b > 4b, but with no change (3b and 4b) or a slight increase in the gap (2b, 2.74 eV). Third, while the calculated Δ(H–L) values were overestimated by approximately 0.3 eV compared to the electrochemical data, these computational predictions agreed with the trends for compounds 2a, 3a, and 4a. It is worth noting that the magnitude of this decrease was also more pronounced in the experimental voltammetry results (Δ(H–L) (4a–1) = −0.9 eV predicted vs. −0.26 V voltammetry). Consistently with the calculations, compounds 2b, 3b, and 4b did not show any significant change in their respective electrochemical gaps. | ||
| Fig. 5 DFT calculated energies and Kohn–Sham orbital representations of the H−2 to L+2 levels of 4a (a) and 4b (b). | ||
Changes in the energy values of the LUMO and HOMO matched our experiments, as the contribution of the substituents depend on their position, see Fig. 5. On one hand, the distribution of the LUMO levels seems always localized in the π orbitals of the perylene core. In contrast, the HOMO level localized in the perylene core for 4b was degenerate with the ones in the 3,5-methoxy phenyl group, with almost a full overlap between HOMO−2, HOMO−1 and HOMO (Fig. 5b). These results provided a successful explanation about the narrow gap obtained in the CV measurements (vide supra). This effect was also in line with the literature for ortho-substituted perylenes with diphenylamine as the donor,18 producing the destabilization of the HOMO to the point of having isoenergetic HOMO to HOMO−3 levels.
Finally, to obtain a reliable picture of the nature of transitions to the excited states, TD-DFT calculations (CAM-B3LYP/6-31G(d,p) in chloroform) were performed and compared with the experimental absorption spectra (detailed in Section 5 of the SI). The TD-DFT showed that the nature of all the transitions remained HOMO–LUMO, as in the PMI 1. Thus, the effect over the transitions can be described as an intramolecular charge transfer, which is in accordance with a slightly lower oscillator strength for compounds 2a (f = 0.803, ε = 31 mM−1 cm−1) and 2b (f = 0.802, ε = 27 mM−1 cm−1), higher for 3a (f = 0.870, ε = 38 mM−1 cm−1) and 3b (f = 0.865, ε = 40 mM−1 cm−1) and intermediate for compounds 4a (f = 0.844, ε = 34 mM−1 cm−1) and 4b (f = 0.836, ε = 34 mM−1 cm−1), as shown in their lower extinction coefficients. The calculated shifts in absorbance also agreed with the stabilization of LUMO levels and matched the experimental results with a bathochromically shifted S0 → S1 transition in 4a > 3a > 2a > 1, and hypsochromically shifted in 3b and 4b.
Conclusions
A family of symmetric ortho-substituted PMIs bearing donor (3,5-dimethoxy-phenyl) or acceptor (3,5-ditrifluoromethyl-phenyl) groups was synthesized and systematically studied. Derivatizations were designed either to preserve or to invert the intrinsic dipole.Crystallographic analysis revealed that polarity and steric effects govern solid-state packing: the dipole-preserving derivative (4a) formed tight slip-stacked arrangements, while the inverted analogue (4b) yielded dimeric cage-like structures with large voids.
Electrochemical and computational studies consistently showed reduced LUMO levels and narrowed HOMO–LUMO gaps when the intrinsic dipole was maintained, along with nearly degenerate HOMO states upon donor substitution at positions 2 and 5. Optical properties mirrored these effects: dipole-preserving derivatives displayed bathochromic shifts without loss of quantum yield, while donor substitution ortho to the imide quenched fluorescence (2b, 4b). In contrast, derivatives with only an acceptor opposite to the dipole (3b) exhibited blue-shifted but highly emissive behavior. Solid-state optical studies together with aggregation experiments further highlighted how molecular packing dictates emission. Broader and red-shifted bands were observed, while the emission was maximized when acceptors were placed ortho to the imide. However, since substitution at positions 2 and 5 of the PMI facilitates aggregation compared to 8 and 11, it also leads to lower fluorescence quantum yields (≈5%) as opposed to the results in solution.
Overall, this work establishes ortho-substituted PMIs as versatile dipolar platforms, where precise donor–acceptor engineering enables control over packing, electronic structure, and photophysics, paving the way for advanced optoelectronic and bio-related applications.42
Author contributions
Investigation: J. G.-C. and M. M. M.; writing – original draft: J. G.-C.; writing – review & editing: J. G.-C., I. R.-M. and T. T.; conceptualization, funding acquisition and project administration: J. G.-C. and T. T.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: detailed experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d5sc06682d.CCDC 2483851 (2a), 2483852 (4a) and 2483853 (4b) contain the supplementary crystallographic data for this paper.43a–c
Raw data of the UV-vis, absorption, fluorescence and electrochemical measurements, as well as the FID files, can be found at an institutional repository with the DOI: https://www.doi.org/10.21950/DJJYER. Further computational output is also available on request.
Acknowledgements
J. G.-C. and T. T. acknowledge financial support from the Spanish MCIN/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR (PID2020-116490GB-I00, TED2021-131255B–C43), MCIU/AEI/10.13039/501100011033/FEDER, UE (PID2023-151167NB-I00), the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM1)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds. J. G.-C. acknowledges his funding from “Ayudas María Zambrano para la atracción de talento internacional” (CA3/RSUE/2021-00243) from the Spanish Government/EU funds at Universidad Autónoma de Madrid, and “Ayudas de atracción de talento de la Comunidad de Madrid 2022” (2022-T1/IND23907) supported by Comunidad de Madrid with EU funds at IMDEA Nanociencia. J. G.-C and T. T. acknowledge support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, grant no. SEV-2016-0686 and CEX2020-001039-S) at IMDEA Nanociencia. We acknowledge the generous allocation of computer time at “Centro de Computación Científica” at the Universidad Autónoma de Madrid (CCC-UAM). We also acknowledge the contribution of the facilities and the members of the Monocrystal DRX lab from the “Servicio Interdepartamental de Investigación” (SIdI) at the Universidad Autónoma de Madrid. We thank Irene Abajo-Cuadrado for the Fluorescence Lifetime measurements and acknowledge the facilities of “Parque científico tecnológico” from Universidad de Burgos for those measurements.References
- G. Horowitz, F. Kouki, P. Spearman, D. Fichou, C. Nogues, X. Pan and F. Garnier, Adv. Mater., 1996, 8, 242–245 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CrossRef CAS.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef CAS.
- P. Chal, A. Shit and A. K. Nandi, ChemistrySelect, 2018, 3, 3993–4003 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chemistry, 2001, 7, 2245–2253 CrossRef.
- L. F. Dössel, V. Kamm, I. A. Howard, F. Laquai, W. Pisula, X. Feng, C. Li, M. Takase, T. Kudernac, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2012, 134, 5876–5886 CrossRef PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- J. Wang, H. Wei, J. Guan, K. Müllen and M. Yin, Chem.–Eur. J., 2025, 31, e202403234 CrossRef CAS PubMed.
- T. C. Lovell, B. P. Branchaud and R. Jasti, Eur. J. Org Chem., 2024, 27, e202301196 CrossRef CAS.
- R. Roy, A. Khan, O. Chatterjee, S. Bhunia and A. L. Koner, Org. Mater., 2021, 3, 417–454 CrossRef CAS.
- I. Papadopoulos, D. Gutiérrez-Moreno, P. M. McCosker, R. Casillas, P. A. Keller, Á. Sastre-Santos, T. Clark, F. Fernández-Lázaro and D. M. Guldi, J. Phys. Chem. A, 2020, 124, 5727–5736 CrossRef CAS PubMed.
- S. Bibi, P. Li and J. Zhang, J. Mater. Chem. A, 2013, 1, 13828 RSC.
- R. Turrisi, A. Sanguineti, M. Sassi, B. Savoie, A. Takai, G. E. Patriarca, M. M. Salamone, R. Ruffo, G. Vaccaro, F. Meinardi, T. J. Marks, A. Facchetti and L. Beverina, J. Mater. Chem. A, 2015, 3, 8045–8054 RSC.
- P. Harrisson, J. Morris, T. B. Marder and P. G. Steel, Org. Lett., 2009, 11, 3586–3589 CrossRef CAS PubMed.
- D. Sánchez-Fernández, T. Torres and J. García-Calvo, J. Org. Chem., 2025, 90, 3202–3208 CrossRef.
- H. Wonneberger, I. Bruder, R. Send, G. Battagliarin, C. Li and K. Mullen, US Pat., 2014 Search PubMed.
- J. Merz, L. Dietrich, J. Nitsch, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem.–Eur. J., 2020, 26, 12050–12059 CrossRef CAS PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS.
- J. R. Palmer, S. B. Tyndall, G. C. Mantel, O. J. Buras, R. M. Young, M. D. Krzyaniak and M. R. Wasielewski, J. Am. Chem. Soc., 2025, jacs5c03973 Search PubMed.
- R. Roy, S. Chawla, V. Sharma, A. K. Pal, Y. Silori, A. Datta, A. K. De and A. L. Koner, Chem. Sci., 2024, 15, 6363–6377 RSC.
- C. Lin, Y. Qi, P. J. Brown, M. L. Williams, J. R. Palmer, M. Myong, X. Zhao, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2023, 14, 2573–2579 CrossRef CAS PubMed.
- P. Rybczyński, M. H. E. Bousquet, A. Kaczmarek-Kędziera, B. Jędrzejewska, D. Jacquemin and B. Ośmiałowski, Chem. Sci., 2022, 13, 13347–13360 RSC.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- T. Miletić, A. Fermi, I. Papadakis, I. Orfanos, N. Karampitsos, A. Avramopoulos, N. Demitri, F. De Leo, S. J. A. Pope, M. G. Papadopoulos, S. Couris and D. Bonifazi, Helv. Chim. Acta, 2017, 100, e1700192 CrossRef.
- J. Sung, P. Kim, B. Fimmel, F. Würthner and D. Kim, Nat. Commun., 2015, 6, 8646 CrossRef CAS.
- F. Marin, S. Tombolesi, T. Salzillo, O. Yaffe and L. Maini, J. Mater. Chem. C, 2022, 10, 8089–8100 RSC.
- A. Dannenhoffer, CCDC 1817935: Experimental Crystal Structure Determination, 2020, DOI:10.5517/ccdc.csd.cc1z0q2s.
- D. Hartmann, S. E. Penty, M. A. Zwijnenburg, R. Pal and T. A. Barendt, Angew. Chem., Int. Ed., 2025, 64, e202501122 CrossRef CAS PubMed.
- M. L. Williams, A. F. Coleman, K. R. Peinkofer, R. M. Young and M. R. Wasielewski, Chem. Sci., 2024, 15, 11472–11479 RSC.
- T. Chand, L. Khamari, D. Chopra, S. Mukherjee and M. Kapur, Chem.–Eur. J., 2022, 28, e202200723 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem. Sci., 2019, 10, 7516–7534 RSC.
- Y. He, C. Mao, M. Duan, L. Fan, X. Wang, Y. Cai, M. Du, M. Hu, P. Hu, Q. Cheng and X. Hu, Org. Chem. Front., 2022, 9, 6466–6474 RSC.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- X. Li, H. Wang, J. A. Schneider, Z. Wei, W.-Y. Lai, W. Huang, F. Wudl and Y. Zheng, J. Mater. Chem. C, 2017, 5, 2781–2785 RSC.
- J. Salbeck, H. Kunkely, H. Langhals, R. W. Saalfrank and J. Daub, CHIMIA, 1989, 43, 6 CrossRef CAS.
- N. Ding, Y.-C. Liao, G. Wang, K.-H. Chang, Z. Wang, K. Liu, J. Ma, P.-T. Chou and Y. Fang, CCS Chem., 2023, 5, 2922–2932 CrossRef CAS.
- B. Schweda, M. Reinfelds, J. Hofinger, G. Bäumel, T. Rath, P. Kaschnitz, R. C. Fischer, M. Flock, H. Amenitsch, M. C. Scharber and G. Trimmel, Chem.–Eur. J., 2022, 28, e202200276 CrossRef CAS PubMed.
- J. García-Calvo, P. Calvo-Gredilla, M. Ibáñez-Llorente, D. C. Romero, J. V. Cuevas, G. García-Herbosa, M. Avella and T. Torroba, J. Mater. Chem. A, 2018, 6, 4416–4423 RSC.
- T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532–2535 CrossRef CAS PubMed.
- A. Brand, L. Allen, M. Altman, M. Hlava and J. Scott, Learn. Publ., 2015, 28, 151–155 CrossRef.
- (a) CCDC 2483851: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pcn7z; (b) CCDC 2483852: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pcn80; (c) CCDC 2483853: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2pcn91.
| This journal is © The Royal Society of Chemistry 2026 |