
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise graphitic nitrogen-incorporation by electrochemical oxidation
Leilei
Xu
a,
Zhibo
Zhang
a,
Hong
Zhou
b,
Ziqi
Wen
c,
Yuxuan
Liu
a,
Heng
Dong
*a and
Wei
Xie
 *b
*b
aMOE Key Laboratory of Pollution Processes and Environmental Criteria, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: dongheng@nankai.edu.cn
bState Key Laboratory of Advanced Chemical Power Sources, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Tianjin Key Lab of Biosensing & Molecular Recognition, Haihe Laboratory of Sustainable Chemical Transformations, Renewable Energy Conversion and Storage Center, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: wei.xie@nankai.edu.cn
cCollege of Materials Science and Engineering, Nankai University, Tianjin 300350, China
First published on 30th December 2025
Abstract
Graphitic nitrogen (graphitic-N) plays a very important role in energy conversion and environmental protection. Although various synthesis methods have been developed, complex devices and harsh conditions are often needed, causing difficulty in flexible regulation. Electrochemical approaches are attracting increasing attention due to their mild reaction conditions, controllability, and environmental compatibility. However, precisely incorporating graphitic-N remains a significant challenge. In this study, a synthesis strategy is designed that creates carbon single vacancies via electrochemical oxidation and then incorporates N radicals to construct graphitic-N. Graphite paper doped with exclusively graphitic-N was achieved by using ammonium ions as the nitrogen source. By integrating multiple operando electrochemical characterization techniques and density functional theory calculations, the crucial regulation parameters were clarified, and the proposed doping mechanism was validated. Hydroxyl radicals generated from electrochemical water dissociation performed three functions: involving the formation of (i) carbon single vacancies and (ii) adjacent oxygen-containing functional groups, and (iii) activation of ammonium ions into N radicals. Ketones exhibited better thermodynamic behavior than hydroxyl species when assembling N radicals into carbon single vacancies. The findings offer both experimental and theoretical foundations for a deeper understanding of the structure–property relationships of graphitic-N and broaden the application prospects of graphitic-N-doped materials.
Introduction
Graphitic-N denotes nitrogen atoms replacing sp2-hybridized carbon atoms in the hexagonal lattice of graphene. This involves typical in-plane nitrogen doping, where the radius of the nitrogen (N) atom (0.74 Å) is close to that of a carbon (C) atom (0.77 Å), thus preserving the coplanarity and conjugated structure of the carbon framework.1–3 Owing to its higher electronegativity (3.04) relative to carbon (2.55), nitrogen doping—particularly in the form of graphitic-N—alters the local electronic environment within the carbon matrix. Graphitic-N, which acts as an electron-donating species, induces an upward shift of the Fermi level.4–6 This electronic modulation enhances interfacial interactions governed by Lewis acid–base sites and π–π stacking, thereby improving the material's reactivity in surface-related processes.7 For example, graphitic-N demonstrates a specific adsorption location for peroxide by forming a metastable intermediate with the carbon plane.8 It paves the way for the development of non-radical advanced oxide processes based on direct electron transfer, exhibiting stronger selectivity for electron-rich emerging contaminants.9–11 In addition, graphitic-N doped carbon cathodes display metal-like catalytic activity for the four-electron oxygen reduction reaction (ORR), and offer superior resistance to methanol crossover.12,13 This shows that carbon materials are promising alternatives for non-metal cathodes in fuel cells.A variety of physico-chemical methods for graphitic-N synthesis have been reported, including chemical vapor deposition,14,15 thermal methods,10,11,16 and plasma methods.17 However, these approaches typically require harsh reaction conditions and complex devices, involving high temperatures, high vacuum, toxic or explosive reagents, and extended processing time. In contrast, electrochemical methods are receiving growing attention, relying on their outstanding advantages in terms of mild reaction conditions, strong controllability and greater potential for industrial production. Common electrochemical techniques such as chronopotentiometry, voltammetry, and chronoamperometry have been adopted to dope nitrogen atoms into graphite-based materials under normal temperature and pressure. Multiple readily available nitrogen sources, including inorganic NH4+/NO3−18–23 and organic compounds (e.g., ionic liquids,24 glycine,25 melamine26), have been employed in those studies.
Although nitrogen doping has been successfully realized in electrochemical systems, mixed co-doping of pyridinic, pyrrolic, and graphitic nitrogen was generally obtained. Graphitic-N is a typical n-type dopant, which can effectively improve the carrier density and conductivity of carbon materials. Therefore, it represents a favorable structure modification for the graphite electrodes used in electrochemical energy-storage applications. However, pyridinic-N and pyrrolic-N act as p-type dopants, which tend to induce carrier scattering and increase resistance, thereby hindering conductivity-oriented device performance. Therefore, this work aims to incorporate graphitic-N into the carbon framework precisely. In addition, in the current research, the synergistic interaction between the electrolysis mode and nitrogen species is still unclear. In particular, real time control over the site-specific evolution mechanisms involved in the graphitic-N formation remains a challenge, thereby limiting the tunability of the graphitic-N content.
Given the apparent structure of graphitic-N, i.e. a nitrogen atom substituting for one sp2-C atom, we designed a controllable synthesis strategy consisting of vacancy-making and nitrogen-filling. Specifically, carbon single vacancies (SVs) are first created on a graphite topological plane via electrochemical oxidation. Then, N radicals are produced by electrochemically activating the nitrogen-containing electrolyte to occupy these SVs. The inorganic ammonium cation is a typical nitrogen source with advantages including low toxicity and good shelf stability, compared with organic amine cations and nitrate anions. In addition to our previous research on electrochemical ammonia oxidation,27,28 in this work, the electrooxidation of (NH4)2SO4 was adopted to obtain N radicals. Crucial electrochemical parameters, which enable modulation of the concentrations of SVs and N radicals, were systematically examined. Advanced operando characterization techniques—including surface-enhanced Raman spectroscopy (SERS), electron paramagnetic resonance (EPR), differential electrochemical mass spectrometry (DEMS), and density functional theory (DFT) calculations—were utilized for comparisons. The results confirmed the feasibility of our strategy, and a sample with 100% relative graphitic-N doping was achieved. The doping mechanism and tuning approach were expounded in detail. This work provides a platform for systematically investigating the structure–property relationships between graphitic-N content and material performance, offering theoretical insights for optimizing catalysts, electrode materials, and sensors.
Compared with traditional graphitic-N doping techniques, such as CVD, high-temperature annealing, and plasma treatment, our electrochemical strategy—consisting of vacancy-making and nitrogen-filling—exhibits notable advantages. This method operates under mild conditions (room temperature and pressure) and requires simple instrumentation and nontoxic reagents. By first generating carbon vacancies and subsequently introducing nitrogen radicals for selective filling, nitrogen atoms can directly substitute sp2-C sites, enabling spatially localized and configuration-oriented doping. This effectively suppresses the co-formation of pyridinic-N and pyrrolic-N, allowing the generation of graphitic-N with a much higher phase purity than conventional approaches. In addition, doping parameters, including potential, current density, pulse design, and electrolyte composition, can be tuned in real time, allowing programmable modulation of vacancy density and nitrogen radical concentration. This offers precise control over both doping level and nitrogen configuration. Furthermore, this electrochemical environment is compatible with operando characterization (e.g. SERS, EPR, DEMS, and DFT), enabling direct tracking of intermediate species and bonding evolution. Such capability strengthens mechanistic understanding and establishes a clearer structure–function correlation. Owing to its universality toward diverse carbon frameworks and scalable process conditions, this strategy shows strong potential for industrial application. In summary, the method provides a precise, green, and scalable route for high-graphitic-N incorporation, offering advantages in both incorporation precision and mechanistic controllability.
Results and discussion
Synthesis and characterization
A strip of graphite paper (1.5 cm × 0.8 cm × 0.02 mm) was used as the working electrode (Fig. 1a), and platinum wire and Ag/AgCl were used as the counter electrode and reference electrode, respectively, to construct a three-electrode cell. In the vacancy-making step, concentrated sulfuric acid (50 wt%) was selected as the electrolyte, and a constant potential of 1.8 V was applied. The oxidation period should be less than 30 min, or else the skeleton of the graphite paper will be damaged and broken into small fragments. Owing to the SO42− intercalation as well as violent H2O dissociation, lots of reactive oxidative radicals immediately grow on the outer and inner surfaces of the graphite layers.29,30 They may contribute to the rapid formation of oxygen-containing functional groups (e.g. hydroxyl –OH, carbonyl –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and carboxyl –COOH) on the plane and edges and even the expected SV.31–33 The sample obtained in this step is labeled as electrochemical graphene oxide (EGO) (Fig. 1b). In the nitrogen-filling step, the electrolyte was replaced by 1.25 M (NH4)2SO4 aqueous solution, and an oxidation potential lower than 1.8 V was applied for 30 min. It is assumed that the nitrogen atom in the NH4+ can be dehydrogenated and precisely filled into the SV as graphitic-N, producing g-ENGO, as shown in Fig. 1c.
O, and carboxyl –COOH) on the plane and edges and even the expected SV.31–33 The sample obtained in this step is labeled as electrochemical graphene oxide (EGO) (Fig. 1b). In the nitrogen-filling step, the electrolyte was replaced by 1.25 M (NH4)2SO4 aqueous solution, and an oxidation potential lower than 1.8 V was applied for 30 min. It is assumed that the nitrogen atom in the NH4+ can be dehydrogenated and precisely filled into the SV as graphitic-N, producing g-ENGO, as shown in Fig. 1c.
 | ||
| Fig. 1 Catalyst preparation process: optical images and structural model of (a) graphite, (b) EGO, and (c) g-ENGO. Atom representations are O (red), N (blue), and C (gray). (d) AFM morphological analysis of graphite, EGO, and g-ENGO. (e) SEM images, and (f and g) TEM images of g-ENGO. (h) Contact angles of graphite, EGO, g-ENGO, and g-ERNGO. | ||
By comparing the optical photos, it can be seen that the shape of the graphite paper remained unchanged, suggesting its convenience when working as an electrode. However, the photos exhibit significant differences in the surface roughness. According to the atomic force microscopy (AFM) measurements (Fig. 1d), the graphite surface was relatively smooth with a roughness (Ra) of ≈0 nm. After electrochemical oxidation, a few “stalactite”-like protrusions appeared on the surface of the EGO, corresponding to an increased roughness (Ra) of ≈300 nm. When the nitrogen atoms were assembled in the g-ENGO, these “stalactites” transformed into “ridge”-like distributions, and the roughness (Ra) increased to ≈500 nm. The surface morphology of the g-ENGO was further observed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. S2). From the SEM images with a magnification of 100 times, obvious wrinkles can be observed, which probably result from the oxygen-containing functional group generation and oxygen bubble evolution (Fig. 1e). Under the TEM view with a magnification of 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times (Fig. 1f), g-ENGO looks like a smooth gauze, which is the typical morphology of a two-dimensional nanocarbon material. Because of the SO42− intercalation, its lattice fringe spacing (0.46 nm) (Fig. 1g) is larger than the normal spacing of few-layer graphene (0.34 nm). A similar phenomenon can also be seen for the graphite paper with a thickness of 1 mm (Fig. S3).
000 times (Fig. 1f), g-ENGO looks like a smooth gauze, which is the typical morphology of a two-dimensional nanocarbon material. Because of the SO42− intercalation, its lattice fringe spacing (0.46 nm) (Fig. 1g) is larger than the normal spacing of few-layer graphene (0.34 nm). A similar phenomenon can also be seen for the graphite paper with a thickness of 1 mm (Fig. S3).
As the surface characteristics of graphite were significantly altered after oxidation and N-doping, the hydrophilicity was correspondingly changed. According to the contact angle testing (Fig. 1h), graphite paper demonstrates the poorest hydrophilicity (contact angle: 89°) because its non-polar surface characteristics weakly interact with polar water molecules. When a substantial number of oxygen-containing functional groups were introduced in the EGO, its surface polarity was significantly enhanced, exhibiting a great enhancement in hydrophilicity with a contact angle of 30°. Meanwhile, the hydrophobicity of the g-ENGO exhibited a slight recovery (contact angle: 45°), suggesting that the graphitic-N assembly may consume adjacent oxygen-containing groups and partially restore the non-polar sp2 conjugated structure. The electrochemical reduction of g-ENGO was performed to further remove some oxygen-containing functional groups (g-ERNGO). As expected, the hydrophobicity increased, with a contact angle of 73°. These results support the concept that the hydrophilicity of g-ENGO can be flexibly adjusted in situ by using the electrochemical method, which usually plays a key role in an aqueous chemical reaction.
Precise regulation of graphitic nitrogen
In the proposed hypothesis, forming SVs is the prerequisite for incorporating graphitic-N, and a series of characterization techniques was adopted to verify their existence. EPR is a visual and quantitative method for determining the concentration of SVs in EGO (Fig. S4). It can be observed from Fig. 2a that the characteristic peak pattern of the SV emerges at a g factor of 2.003, and the content gradually increases with the oxidation time.34 In particular, in the first 10 min, the SV content increased from 0 to 1.493, indicating relatively fast oxidation kinetics. As the SV theoretically stems from carbon emission, operando DEMS in linear sweep voltammetry (LSV) mode for ten cycles was carried out to monitor and identify the gaseous carbon molecules being released from the graphite planes. As shown in Fig. 2b, the signals with specific mass charge ratios (m/z) of 28 (CO) and 44 (CO2) appeared repeatedly and stably in each cycle. In addition, the deconvoluted C1s and O1s XPS spectra display three types of oxygen-containing functional groups: hydroxyl (–OH), carbonyl (–CO) and carboxyl groups (–COOH) (Fig. S5b and 5c). Referring to the results described above, we predict that the formation of SVs is accompanied by the detachment of –OH and –CO, which is due to continuous electrochemical oxidation at 1.8 V. The C/O ratios and high-resolution O1s spectra obtained from XPS measurements (Fig. S5a) for EGO-5, EGO-10, EGO-20, and EGO-30 provide direct evidence for the prediction. As the electrochemical oxidation proceeds, the C/O ratios fluctuate up and down, and do not exhibit the linear-like decreasing trend observed in the electrochemical exfoliation of GO.30
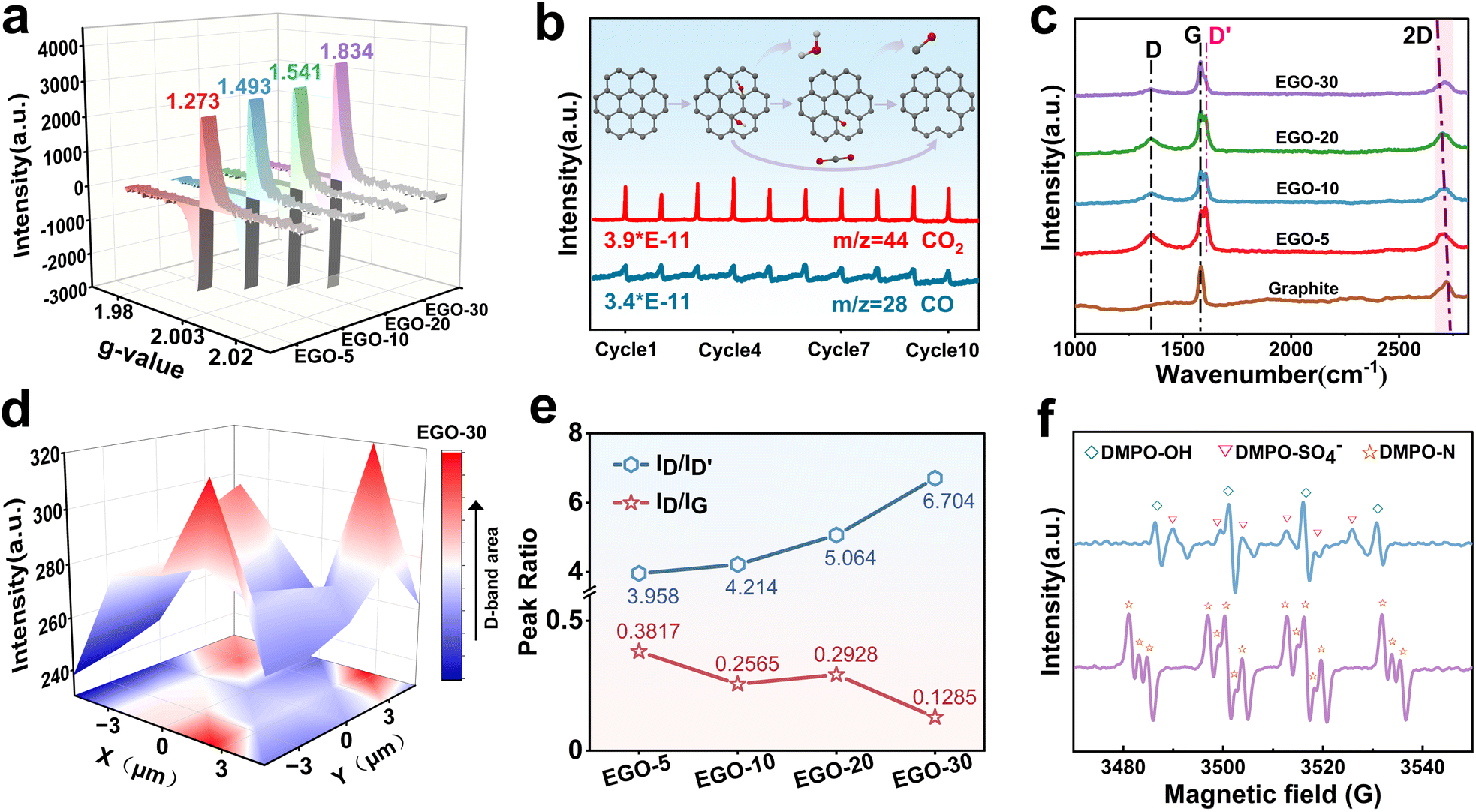 | ||
| Fig. 2 (a) EPR spectra of EGO. (b) Operando DEMS results for EGO. Atom representations are O (red), C (gray), and H (white). (c) Raman spectra. (d) D-band Raman mapping for EGO. (e) ID/ID′ and ID/IG change curves. (f) Operando EPR spectra of DMPO-OH, DMPO-SO4−, and DMPO-N during electrochemical processes. | ||
Raman spectroscopy analysis (Fig. 2c) was further performed to characterize the carbon structure of the EGO. The overall ID/IG ratio shows a downward trend, indicating that as the oxidation time was extended, sp3 carbon decreased and SVs increased. Compared to the graphite, the 2D band (2720 cm−1) of the EGO undergoes a red shift, which also suggests the transformation of in-plane sp2 carbon to distorted sp3 carbon and the development of defects.35 In addition to the typical D-band (1350 cm−1) and G band (1580 cm−1) peaks, the characteristic peak for the D′ band was observed at 1611 cm−1 for EGO. The intensity of the D′ band, originating from a double resonance Raman feature induced by disorder and defects, has been reported to be a measure for distinguishing boundary-like defects (ID/ID′ = ∼3.5), vacancy-type defects (ID/ID′ = ∼7), and sp3-type defects (ID/ID′ = ∼13) by comparing with the intensity of the D-band. Herein, the ID/ID′ ratio increased with the oxidation duration from 3.958 (EGO-5) to 6.704 (EGO-30), indicating that the boundary-like defects gradually transformed into vacancy-type defects (Fig. 2e).36 Meanwhile, 3D Raman mapping of the D peak from the EGO-30 within a square area of 10 µm × 10 µm displays uniform distribution of the vacancies on the graphite plane (Fig. 2d).
Based on these results, it can be confirmed that SVs were successfully produced in the EGO, and their concentration can be adjusted by controlling the oxidation time.
Electrochemically activating NH4+ to N radicals is the second step to form graphitic-N. The previous studies have speculated that hydroxyl radicals can attack ammonium ions to oxidize them into N radicals in an electrochemical system.37,38 In order to explore this using experiments, operando electrochemical EPR measurement was performed in LSV mode, and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was used as the trapping agent. As shown in Fig. 2f, EPR signals from DMPO-OH, DMPO-SO4−, and DMPO-N spin adducts all appeared, indicating the formation of hydroxyl radicals, sulfate radicals, and N radicals (*NH2 and/or *NH).39,40
Precisely doping graphitic-N with a controllable concentration is an anticipated advantage of the electrochemical method. Based on the prepared EGO at 1.8 V with different oxidation times (0–30 min), a series of g-ENGO samples was prepared under a constant 1.6 V. From the deconvoluted N1s XPS spectrum (Fig. 3a and S7), the doped N states are composed of graphitic-N (401.6 eV) and pyrrolic-N (399.9 eV), with no pyridinic-N detected (398.8 eV).16 This represents a significant difference compared to the chemical methods. By exploring the relationship between the carbon vacancy content and the relative graphitic-N content (Fig. 3c), it was found that as the carbon vacancy content increases, the relative graphitic-N content exhibits a parabolic trend. The highest relative graphitic-N content of 73% was obtained for g-ENGO-20. At this point, the absolute graphitic-N content reaches 1.581% (Fig. S9). This is because an excessive oxidation duration will transform a carbon single vacancy into a double-vacancy or even hole defects, preventing nitrogen atoms from tridentate coordination.32 Clearly, the presence of SVs is an essential condition for the formation of graphitic-N, which perfectly validates our hypothesis. Controlling the oxidation time of the EGO is one of the effective methods to adjust the graphitic-N content. The pioneers of this work believed that pyrrolic-N is formed via the nucleophilic attack of N-containing species on the carbonyl functional groups.41 Therefore, the lowest pyrrolic content of 26% in g-ENGO-20 can be attributed to its lowest amount of carboxyl functional groups (∼0%) (Fig. S5). It can also be noted that even the graphite paper was doped by electrochemical oxidation without pre-oxidation (g-ENGO-0), and graphitic-N still emerged, which confirms the effect of water dissociation on SV generation. The negative results obtained from the methyl-added system also indicate the important effect of water dissociation (Fig. S11 and Table S3).
 | ||
| Fig. 3 (a) High-resolution N1s XPS of g-ENGO-x (x represents different oxidation times). (b) High-resolution N1s XPS of g-ENGO (at different doping potentials). (c) The relationship between carbon vacancy concentration and relative graphitic-N content as a function of oxidation time. (d) The relationship between nitrogen radical concentration and relative graphitic-N content as a function of voltage. (e) Raman spectra. (f) Plots of work functions and potential maps of graphite, EGO, and g-ENGO samples derived from KPFM results (inset). | ||
We predict that anchoring a nitrogen atom into the carbon via the N radical is another crucial process for constructing graphitic-N. When the distribution of the SVs is determined, the N radical production is probably an adjustable parameter to control the concentration of graphitic-N in the g-ENGO. As the oxidation potential has a significant influence on this process (Fig. S6), a series of doping potentials (0.8 V to 1.8 V) for the g-ENGO was examined with EGO-30 as the basic sample (Fig. 3b and S8). Similar to the influence of the oxidation time during EGO synthesis, the relative graphitic-N content also exhibited a parabolic trend with the doping potential (Fig. 3d and S10). Specifically, 100% relative content of graphitic-N was obtained under 1.2 V, indicating that N radicals play a key role in the formation of graphitic-N. A quantitative comparison with prior work, including synthesis parameters, total nitrogen content, and nitrogen species distribution, is provided in Table S1. The advantage of our electrochemical method for the precise incorporation of graphitic-N can be clearly understood. According to the assumption from the contact angle test that oxygen-containing functional groups may be involved in graphitic-N formation, we conducted a nitrogen-doping experiment by pyrolyzing SV-contained graphite and melamine at 900 °C (Fig. S12). Due to the lack of oxygen-containing functional groups on the graphite plane, no nitrogen atoms were doped in. This suggests that oxygen-containing functional groups are the third essential participants for graphitic-N assembly, especially the adjacent ones at the SVs.
The incorporation of graphitic-N on the graphite plane was further validated via Raman spectroscopy (Fig. 3e) and Kelvin probe force microscopy (KPFM) using the series of g-ENGO adjusted by oxidation time (Fig. 3f). Compared with the original graphite, the ID/IG ratio of the g-ENGO increased significantly, confirming the gradual formation of defect structures due to nitrogen doping. In addition, the G band (1580 cm−1) of the g-ENGO exhibits a blue shift, which is a typical demonstration of nitrogen doping in graphite.42 Meanwhile, KPFM work function measurements show opposite regulatory effects of vacancy doping and graphitic-N doping on the Fermi level of graphene. Using the work function (Φ ≈ 5.03 eV) of the graphite as a reference, EGO gives a higher work function of ∼5.26 eV, indicating a downward shift in the Fermi level. In contrast to that, g-ENGO-1.2 gives a lower work function of ∼4.82 eV, indicating an upward shift in the Fermi level.43
Synthesis mechanism
Based on the experimental results, the proposed mechanism regarding nitrogen transformation from ammonia ions to N radicals was further verified by SERS (Fig. 4a and S13). Detailed experimental procedures are provided in the SI. By performing LSV over a potential range of 0.8 V to 1.95 V (vs. Ag/AgCl), we successfully detected the stretching vibration modes of *NH2 (410 cm−1), *NH (662 cm−1), and *NNH (585 cm−1), which are key intermediates needed in the graphitic-N doping process.27,28 Additionally, the stretching vibration peak of the active oxygen radical (*O) was consistently observed at 516 cm−1. Within the voltage range of 0.8 V to 1.7 V, the strong intensities of the *NH2 and *O peaks provide strong evidence that the primary N radicals are *NH2, and they are achieved depending on the attack of the hydroxyl radicals on the ammonium ions. When the potential continually increases to 1.9 V, the intensity of the *O peak decreases and the *NH2 peak disappears, indicating that excessive potential will cause vigorous hydrolysis, resulting in the undesirable production of oxygen. Peaks corresponding to *NH are also observed in the 0.8 V to 1.7 V range, and their intensities continuously increase with the potential. The similar trends for *NH2 and *NH indicate that the adsorbed *NH2 undergoes a dehydrogenation reaction on the EGO surface to form *NH. Additionally, the *NNH peak becomes prominent in the range of 1.4 V to 1.7 V, but nearly disappears when the potential continues to rise. It indicates that a significant portion of the adsorbed *NH undergoes N–N coupling reactions to form N2.28 | ||
| Fig. 4 (a) SERS spectra of ammonia dehydrogenation in an aqueous solution of 1.25 M (NH4)2SO4. (b) Fragmental structures for the stationary points involved in route-a, route-b, and route-c. Atom representations are O (red), N (blue), C (gray), and H (white). Other carbon atoms are omitted for clarity. The distances are represented in units of angstroms. Relative energies (in parentheses) are given in kcal mol−1. (c) Reaction energy distribution of ammonia-mediated a-INT1 (route-a in black) and bc-INT1 (route-b in blue and route-c in red) doping reactions. | ||
DFT calculations were performed to clarify the reaction mechanism from EGO to g-ENGO from a thermodynamic perspective (Fig. 4b and c). Given the experimental results above, we constructed two EGO models: a single carbon vacancy on a finite graphene flake with one carbonyl (–CO) group (EGO-1) or adjacent two hydroxyl (–OH) groups (EGO-2) (Fig. S14). The three unsaturated carbon atoms surrounding the vacancy were labeled as C1, C2, and C3 (Fig. 4b and S15). Route-a was exhibited for graphitic-N doping in EGO-1, with localized amplification of a-INT1. The carbonyl group is bonded to C1, and an external –NH2 group adsorbs onto the carbonyl site with a binding energy of 3.03 kcal mol−1. Subsequently, the nitrogen atom bonds with C2, followed by the transfer of one hydrogen atom from –NH2 to the oxygen atom, forming an –OH group. The nitrogen then bonds with C3, resulting in two new C–N bonds on the carbon surface. Finally, the nitrogen forms a bond with C1, undergoing a second N–H dissociation (ab-INT2). The released hydrogen atom attacks the oxygen atom, forming an H2O molecule and simultaneously completing the formation of graphitic-N (abc-INT2). Route-b and route-c were both calculated for graphitic-N doping in EGO-2 (bc-INT1). The two hydroxyl groups are bonded to C1 and C3, and the –NH2 group adsorbs onto them with a binding energy of 3.39 kcal mol−1. The nitrogen atom sequentially bonds with C2 and C1, forming two C–N bonds and two –OH groups. The route bifurcates based on which oxygen atom is attacked first by the hydrogen in –NH2. In route-b, the nitrogen bonds with C3, and the hydrogen attacks the oxygen on C3 to form an H2O molecule. A second N–H cleavage then occurs, with hydrogen attacking the oxygen on C1, generating a second H2O molecule and resulting in graphitic-N formation (abc-INT2). In route-c, the hydrogen atom first attacks the oxygen on C1, forming H2O, followed by nitrogen bonding with C3. A second N–H cleavage yields another H2O molecule through attack on the oxygen at C3, again resulting in graphitic-N.
As shown in Fig. 5c, in all three routes (a, b, and c), the attachment of the –NH2 group to the carbon atom (a-TS1 and bc-TS1) is the rate-determining step, with activation energies of 18.08 kcal mol−1 for route-a and 20.94 kcal mol−1 for routes b and c. These values suggest that graphitic-N doping is kinetically accessible in all three cases. The calculated reaction energies are −149.68 kcal mol−1 for route-a and −179.22 kcal mol−1 for routes b and c, confirming that the graphitic-N deriving from the SV, –NH2, and adjacent oxygen-containing functional groups is thermodynamically favorable under electrochemical conditions.
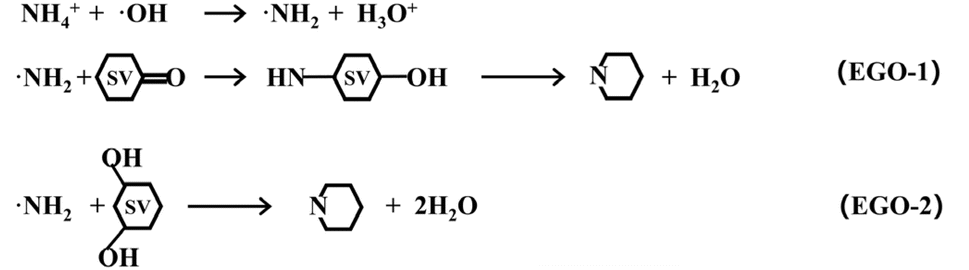
Based on the experimental research and DFT calculations mentioned above, schematic equations to elucidate the mechanism for NH4+ ion conversion into graphitic nitrogen under our electrochemical conditions are provided as follows.
g-ENGO applications
In our previous research, the experiments and DFT calculations clearly showed that peroxydisulfate (PDS) can preferentially adsorb on graphitic-N (−2.46 eV on graphitic-N, −1.30 eV on pyridinic-N, and −1.34 eV on N vacancies).32 This means that electron transfer can occur from graphitic-N to the PDS without any external energy input. This can be attributed to the increased Fermi level of the carbon material by the graphitic-N doping (proved by KPFM here). Therefore, we performed open-circuit potential (OCP) measurement for detecting the electron transfer from graphitic-N to PDS as a reference to evaluate the performance of the g-ENGO obtained using different doping potentials (Fig. 5a). To avoid the interference of the oxygen-containing functional groups, the g-ENGO was reduced at −1 V for 30 min (Fig. S16). The initial OCP of g-ERNGO remained stable, and upon injection of 1 mM PDS at 300 s, the OCP of the g-ERNGO immediately increased by 0.2 V to 0.5 V. Notably, the amplification of the OCP was positively correlated with the relative content of graphitic-N. This result gives a reliable prediction that g-ERNGO can be used as an electrochemical sensor for PDS. | ||
| Fig. 5 (a) Open-circuit potential curves with the addition of 1 mM PDS on g-ENGO-X (X represents the doping potential). (b) LSV curves of g-ENGO-X in 1 M KOH solution, showing the OER catalytic activity of the electrode. (c) Operando EPR spectra of DMPO-N during g-ENGO preparation. | ||
OER performance was also examined as a demonstration of the graphitic-N doping. LSV tests (Fig. 5b) show that the initial overpotentials of g-ERNGO-1.6 V, 1.4 V, 1.2 V, 1 V, and 0.8 V are 1.39 V, 1.41 V, 1.45 V, 1.51 V, and 1.63 V, respectively, significantly lower than that of graphite (1.84 V relative to RHE). In particular, the increase of the absolute graphitic-N content has an obvious role in negatively shifting the overpotential of the OER. Good stability of the g-ERNGO was demonstrated through consecutive cyclic voltammetry (CV) scans for 50 cycles, indicating its application potential in practice (Fig. S18).
As the active oxide radicals derived from OER are crucial for N radical generation during g-ENGO synthesis, we designed a masking experiment by using the operando EPR measurement (Fig. 5c) to verify the active role of graphitic-N in the OER, in which EGO-5, EGO-10, EGO-20, and EGO-30 were used as the working electrode, and graphitic-N doping was performed by oxidizing them for 10 min in 1.25 M (NH4)2SO4. At the same time, the scavenger DMPO is added to the scavenging system to capture N radicals that are not used for doping. Theoretically, the peak intensity of the N radicals should decrease, resulting from the capture by SVs in the graphite. However, as the content of SVs gradually increased from EGO-5 to EGO-30 (Fig. 2a), the concentration of the captured N radicals also exhibited an increasing trend. The reason for this may be that the graphitic-N generated during doping reduces the energy barrier for hydrolysis to hydroxyl groups, thereby promoting the accelerated production of N radicals. This is consistent with our predicted conclusion.
Conclusions
In this work, we proposed a controllable and easy-to-operate electrochemical method enabling the selective synthesis of graphitic-N at ambient temperature and pressure. The sequence process, including vacancy engineering and N radical assembly, allows precise regulation of the graphitic-N content, and 100% relative content of graphitic-N was achieved. By integrating advanced characterization and DFT calculation, we elucidated the formation mechanism of graphitic-N and identified the crucial parameters for its precise regulation. The SVs, N radicals, and adjacent oxygen-containing functional groups are the three essential participants in graphitic-N doping, which significantly depend on electrochemical water dissociation to hydroxyl radicals. This study provides a flexible framework for the systematic preparation of nitrogen-doped carbon materials and paves the way for their application in electrochemical energy conversion (e.g. fuel cells and metal–air batteries), environmental remediation (e.g. advanced oxidation processes and pollutant adsorption), electrochemical sensing, etc., thereby contributing to the development of sustainable, green, and low-carbon technologies.Author contributions
Conceptualization: Leilei Xu, Heng Dong; investigation: Leilei Xu, Zhibo Zhang, Hong Zhou, Ziqi Wen; supervision: Heng Dong, Wei Xie; writing – original draft: Leilei Xu; writing – review & editing: Heng Dong, Wei Xie, Yuxuan Liu.Conflicts of interest
The authors declare no competing interests.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details (materials and characterization); synthesis methods; TEM images, XRD pattern, XPS analysis, results of EPR, construction of models and computation details. See DOI: https://doi.org/10.1039/d5sc08053c.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (U24A20518 & 22574082), the Fundamental Research Funds for the Central Universities, Entrepreneurship Shenzhen Science and Technology Program (KCXFZ20211020172542001) and the 111 project (B25010).Notes and references
- P. Błoński, J. Tuček, Z. Sofer, V. Mazánek, M. Petr, M. Pumera, M. Otyepka and R. Zbořil, Doping with Graphitic Nitrogen Triggers Ferromagnetism in Graphene, J. Am. Chem. Soc., 2017, 139, 3171–3180 CrossRef PubMed.
- Y.-F. Lu, S.-T. Lo, J.-C. Lin, W. Zhang, J.-Y. Lu, F.-H. Liu, C.-M. Tseng, Y.-H. Lee, C.-T. Liang and L.-J. Li, Nitrogen-Doped Graphene Sheets Grown by Chemical Vapor Deposition: Synthesis and Influence of Nitrogen Impurities on Carrier Transport, ACS Nano, 2013, 7, 6522–6532 Search PubMed.
- K. Yokwana, B. Ntsendwana, E. N. Nxumalo and S. D. Mhlanga, Recent advances in nitrogen-doped graphene oxide nanomaterials: Synthesis and applications in energy storage, sensor electrochemical applications and water treatment, J. Mater. Res., 2023, 38, 3239–3263 Search PubMed.
- M. Fan, Z.-Q. Feng, C. Zhu, X. Chen, C. Chen, J. Yang and D. Sun, Recent progress in 2D or 3D N-doped graphene synthesis and the characterizations, properties, and modulations of N species, J. Mater. Sci., 2016, 51, 10323–10349 Search PubMed.
- X. Ning, Y. Li, J. Ming, Q. Wang, H. Wang, Y. Cao, F. Peng, Y. Yang and H. Yu, Electronic synergism of pyridinic- and graphitic-nitrogen on N-doped carbons for the oxygen reduction reaction, Chem. Sci., 2019, 10, 1589–1596 Search PubMed.
- E. B. Yutomo, F. A. Noor and T. Winata, Effect of the number of nitrogen dopants on the electronic and magnetic properties of graphitic and pyridinic N-doped graphene – a density-functional study, RSC Adv., 2021, 11, 18371–18380 Search PubMed.
- B. Başer, B. Yousaf, U. Yetis, Q. Abbas, E. E. Kwon, S. Wang, N. S. Bolan and J. Rinklebe, Formation of nitrogen functionalities in biochar materials and their role in the mitigation of hazardous emerging organic pollutants from wastewater, J. Hazard. Mater., 2021, 416, 126131 Search PubMed.
- Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. S. Su, J. Wang, X. Bao and D. Ma, Nitrogen-Doped sp2-Hybridized Carbon as a Superior Catalyst for Selective Oxidation, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 Search PubMed.
- Y.-X. Huang, L.-Q. Yu, K.-Y. Chen, H. Wang, S.-Y. Zhao, B.-C. Huang and R.-C. Jin, Biochar with self-doped N to activate peroxymonosulfate for bisphenol-A degradation via electron transfer mechanism: The active edge graphitic N site, Chin. Chem. Lett., 2024, 35, 109437 CrossRef CAS.
- F. Li, L. Sun, H. Wang, H. Dong, F. Li, X. Wu and S. Zhan, Crucial role of nitrogen vacancies in carbon activated persulfate toward nonradical abatement of micropollutants from secondary effluent, Appl. Catal. B: Env., 2025, 361, 124584 Search PubMed.
- S. Zhu, X. Huang, F. Ma, L. Wang, X. Duan and S. Wang, Catalytic Removal of Aqueous Contaminants on N-Doped Graphitic Biochars: Inherent Roles of Adsorption and Nonradical Mechanisms, Environ. Sci. Technol., 2018, 52, 8649–8658 Search PubMed.
- T. Jeong, K. Kim, B. H. Kim, S. I. Choi, C. H. Choi, J. Kang and M. Kim, Ligand Engineering of Co-N4 Single-Atom Catalysts for Highly-Active and Stable Acidic Oxygen Evolution, Adv. Sci., 2025, 12, e2502230 Search PubMed.
- G. Murdachaew and K. Laasonen, Oxygen Evolution Reaction on Nitrogen-Doped Defective Carbon Nanotubes and Graphene, J. Phys. Chem. C, 2018, 122, 25882–25892 Search PubMed.
- B. P. Jaisi, R. Zhu, G. Kalita and M. Umeno, Morphological changes of carbon thin films with nitrogen doping synthesized by microwave-excited surface wave plasma CVD, Mater. Chem. Phys., 2023, 307, 128183 CrossRef CAS.
- L. Lin, J. Y. Li, Q. H. Yuan, Q. C. Li, J. C. Zhang, L. Z. Sun, D. R. Rui, Z. L. Chen, K. C. Jia, M. Z. Wang, Y. F. Zhang, M. H. Rummeli, N. Kang, H. Q. Xu, F. Ding, H. L. Peng and Z. F. Liu, Nitrogen cluster doping for high-mobility/conductivity graphene films with millimeter-sized domains, Sci. Adv., 2019, 5, 9 Search PubMed.
- H. Kammoun, B. D. Ossonon and A. C. Tavares, Nitrogen-Doped Graphene Materials with High Electrical Conductivity Produced by Electrochemical Exfoliation of Graphite Foil, Nanomater., 2024, 14, 123 Search PubMed.
- A. Bident, N. Caillault, F. Delange, C. Labrugere, G. Aubert, C. Aymonier, E. Durand, A. Demourgues, Y. Lu and J. F. Silvain, Nitrogen Radiofrequency Plasma Treatment of Graphene, ChemistrySelect, 2023, 8, e202303661 Search PubMed.
- M. B. Arvas, M. Gencten and Y. Sahin, One-step synthesized N-doped graphene-based electrode materials for supercapacitor applications, Ionics, 2021, 27, 2241–2256 Search PubMed.
- R. Bhaskaran and R. Chetty, One-Pot Room Temperature Synthesis of Nitrogen-Doped Graphene and Its Application as Catalyst Support for ORR in PEMFCs, ACS Appl. Energy Mater., 2024, 7, 390–402 Search PubMed.
- F. Lou, M. E. M. Buan, N. Muthuswamy, J. C. Walmsley, M. Rønning and D. Chen, One-step electrochemical synthesis of tunable nitrogen-doped graphene, J. Mater. Chem. A, 2016, 4, 1233–1243 Search PubMed.
- X. Lu and C. Zhao, Controlled electrochemical intercalation, exfoliation and in situ nitrogen doping of graphite in nitrate-based protic ionic liquids, Phys. Chem. Chem. Phys., 2013, 15, 20005–20009 Search PubMed.
- L. Magerusan, F. Pogacean and S. Pruneanu, Enhanced Acetaminophen Electrochemical Sensing Based on Nitrogen-Doped Graphene, Int. J. Mol. Sci., 2022, 23, 14866 Search PubMed.
- A. Shayesteh Zeraati, S. A. Mirkhani, F. Sharif, A. Akbari, E. P. L. Roberts and U. Sundararaj, Electrochemically Exfoliated Graphite Nanosheet Films for Electromagnetic Interference Shields, ACS Appl. Nano Mater., 2021, 4, 7221–7233 Search PubMed.
- X. Sun, N. Yang, H. Dong, H. Yu, H. Yu and L. Feng, In-situ electrochemical synthesis of heteroatoms-doped reduced graphene oxide toward nonradical degradation of tetracycline, Chem. Eng. J., 2023, 471, 144834 Search PubMed.
- Y. Yang, W. Shi, R. Zhang, C. Luan, Q. Zeng, C. Wang, S. Li, Z. Huang, H. Liao and X. Ji, Electrochemical Exfoliation of Graphite into Nitrogen-doped Graphene in Glycine Solution and its Energy Storage Properties, Electrochim. Acta, 2016, 204, 100–107 Search PubMed.
- F. Liu, F. Niu, T. Chen, J. Han, Z. Liu, W. Yang, Y. Xu and J. Liu, One-step electrochemical strategy for in-situ synthesis of S,N-codoped graphene as metal-free catalyst for oxygen reduction reaction, Carbon, 2018, 134, 316–325 CrossRef CAS.
- M. Zhang, T. Wang, X. Du, Y. Mao, A. Zhu, A. Du, L. Tong and W. Xie, Observation of *N2H3 Intermediates by In Situ Electrochemical SERS: New Pathway of Ammonia Oxidation on High-Durability PtIr Catalysts, J. Am. Chem. Soc., 2025, 147, 28331–28339 Search PubMed.
- X. Du, A. Du, D. Wang, Y. Mao, Z. Zhang and W. Xie, Surface-Enhanced Raman Spectroscopic Study of Key Intermediates in Electrochemical Ammonia Decomposition, J. Am. Chem. Soc., 2024, 147, 8083–8087 CrossRef PubMed.
- D. Chen, Z. Lin, M. M. Sartin, T.-X. Huang, J. Liu, Q. Zhang, L. Han, J.-F. Li, Z.-Q. Tian and D. Zhan, Photosynergetic Electrochemical Synthesis of Graphene Oxide, J. Am. Chem. Soc., 2020, 142, 6516–6520 Search PubMed.
- S. Pei, Q. Wei, K. Huang, H.-M. Cheng and W. Ren, Green synthesis of graphene oxide by seconds timescale water electrolytic oxidation, Nat. Commun., 2018, 9, 145 Search PubMed.
- S. Fang, Y. Lin and Y. H. Hu, Recent Advances in Green, Safe, and Fast Production of Graphene Oxide via Electrochemical Approaches, ACS Sustain. Chem. Eng., 2019, 7, 12671–12681 Search PubMed.
- P. Feicht and S. Eigler, Defects in Graphene Oxide as Structural Motifs, ChemNanoMat, 2018, 4, 244–252 Search PubMed.
- S. Yang, S. Brüller, Z.-S. Wu, Z. Liu, K. Parvez, R. Dong, F. Richard, P. Samorì, X. Feng and K. Müllen, Organic Radical-Assisted Electrochemical Exfoliation for the Scalable Production of High-Quality Graphene, J. Am. Chem. Soc., 2015, 137, 13927–13932 Search PubMed.
- C. Jiang, Z. Chen, R. Yang, Z. Luogu, Q. Ren, H. Hu, K. Wang, S. Li, C. Deng, M. Li and L. Zheng, Carbon-Based Flexible Electrode for Efficient Electrochemical Generation of Reactive Chlorine Species in Tumor Therapy, Adv. Healthcare Mater., 2025, 14, 2500369 Search PubMed.
- D. Belotcerkovtceva, R. P. Maciel, E. Berggren, R. Maddu, T. Sarkar, Y. O. Kvashnin, D. Thonig, A. Lindblad, O. Eriksson and M. V. Kamalakar, Insights and Implications of Intricate Surface Charge Transfer and sp3-Defects in Graphene/Metal Oxide Interfaces, ACS Appl. Mater. Interfaces, 2022, 14, 36209–36216 CrossRef CAS PubMed.
- Y. Yamada, K. Murota, R. Fujita, J. Kim, A. Watanabe, M. Nakamura, S. Sato, K. Hata, P. Ercius, J. Ciston, C. Y. Song, K. Kim, W. Regan, W. Gannett and A. Zettl, Subnanometer Vacancy Defects Introduced on Graphene by Oxygen Gas, J. Am. Chem. Soc., 2014, 136, 2232–2235 Search PubMed.
- J. Fan, L. Xie, X. Zhang, P. Zou, Y. Zheng, Y. Fan, H. Wang, X. Jiang and Y. Chang, Mechanically Induced Green Targeted Conversion of Ammonia Nitrogen to N2: Based on Cavitation Effects and ROS Oxidation, Environ. Sci. Technol., 2024, 58, 21569–21577 CrossRef CAS PubMed.
- R. Halseid, J. S. Wainright, R. F. Savinell and R. Tunold, Oxidation of ammonium on platinum in acidic solutions, J. Electrochem. Soc., 2007, 154, B263–B270 Search PubMed.
- J. Dou, Y. Tang, Z. Lu, G. He, J. Xu and Y. He, Neglected but Efficient Electron Utilization Driven by Biochar-Coactivated Phenols and Peroxydisulfate: Polyphenol Accumulation Rather than Mineralization, Environ. Sci. Technol., 2023, 57, 5703–5713 Search PubMed.
- Q. Shi, W. Tang, K. Kong, X. Liu, Y. Wang and H. Duan, Electrocatalytic Upgrading of Plastic and Biomass-Derived Polyols to Formamide under Ambient Conditions, Angew. Chem., Int. Ed., 2024, 63, e202407580 CrossRef CAS PubMed.
- X. R. Wang, X. L. Li, L. Zhang, Y. Yoon, P. K. Weber, H. L. Wang, J. Guo and H. J. Dai, N-Doping of Graphene Through Electrothermal Reactions with Ammonia, Science, 2009, 324, 768–771 Search PubMed.
- R. Arunpandian, M. Kumar, S. I. Lasalle B, P. Vijayakumar and J.-H. Chang, Hierarchical synthesis of multi-layer graphene-like and nitrogen-doped graphitized carbon from dead leaf biomass for high-performance energy storage and CO2 capture, J. Taiwan Inst. Chem. Eng., 2025, 172, 106100 CrossRef CAS.
- P. Szroeder, A. Banaszak-Piechowska and I. Sahalianov, Tailoring Electrocatalytic Properties of sp2-Bonded Carbon Nanoforms Through Doping, Mol., 2025, 30, 1265 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |