
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyl-oxyallenes as α,β-unsaturated ketone surrogates for Giese radical addition
Jiarong
Jin
a,
Xin
Li
a,
Yicheng
Luo
a,
Jianfu
Chen
*c,
Wenjun
Tang
 *ab and
Kang
Du
*a
*ab and
Kang
Du
*a
aSchool of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, Chin. E-mail: kangdu@ucas.ac.cn
bState Key Laboratory of Bio-Organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Ling Ling Rd, Shanghai 200032, China. E-mail: tangwenjun@sioc.ac.cn
cState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, Key Laboratory for Advanced Materials, Centre for Computational Chemistry and Research Institute of Industrial Catalysis, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: jfchen@ecust.edu.cn
First published on 25th November 2025
Abstract
Oxyallenes are valuable building blocks in organic synthesis, most commonly exploited as π-allyl metal precursors in transition-metal-catalyzed allylation reactions. In contrast, their engagement in radical processes remains largely unexplored. Herein, we disclose a Giese-type radical addition protocol in which acyl-substituted oxyallenes function as in situ precursors to α,β-unsaturated ketones, enabling efficient coupling with 2-azaallyl radicals. This metal-free method delivers a wide range of γ-amino ketones in high yields with broad functional group tolerance, mild conditions, and scalability to gram quantities. Mechanistic studies, including radical trapping and isotopic labeling, support a pathway involving radical addition of the 2-azaallyl radical to transient enone intermediates. These findings establish a new reactivity mode of oxyallenes in radical chemistry and provide an efficient route to synthetically and pharmaceutically valuable amino ketones.
Introduction
Oxyallenes represent a versatile class of C3 synthons in modern organic synthesis.1 The presence of an alkoxy group on the allene core induces polarization across the π-system, thereby enabling distinct reactivity profiles at different carbon centers. Under metalation conditions, deprotonation at the C1 position furnishes lithiated alkoxyallenes that react with a wide array of electrophiles.1a–f Conversely, the C2 carbon is susceptible to direct electrophilic attack2a–c or acid-mediated activation—either by Lewis acids1g,h,2d or Brønsted acids2e,f—leading to the formation of reactive oxonium intermediates. These intermediates subsequently undergo nucleophilic additions at the C1 or C3 positions, as illustrated in Fig. 1A. In transition-metal-catalyzed allylation chemistry, oxyallenes readily undergo hydrometallation with a transient M–H species to furnish π-allyl metal intermediates with diverse reactivity profiles. Advances in this area include Pd- and Rh-catalyzed allylic substitution with pronucleophiles,3 and hydrofunctionalization reactions catalyzed by Ir, Ru, and Cu with carbonyls/imines (Fig. 1B).4 Despite their broad utility in polar reactions and transition metal catalysis, radical-based transformations remain conspicuously underexplored.5 Herein, we demonstrate the first use of acyl-substituted oxyallenes as α,β-unsaturated ketone surrogates in Giese-type radical addition reactions. This method allows for the gradual liberation of the enone from acyl-oxyallene while maintaining a relatively lower concentration of enone. This approach offers potential advantages in modulating the molecular weight and distribution of polymers in radical polymerization. Additionally, it could influence both the reactivity and selectivity in the Giese radical addition. | ||
| Fig. 1 Introduction. | ||
As postulated in Fig. 1C, O-acyl oxyallenes can presumably be accessed analogously to alkoxyallenes via base-promoted isomerization of propargylic esters (e.g., MOt-Bu). Unlike relatively stable alkoxyallene, this O-acyl oxyallene intermediate could further undergo a cascade sequence involving transesterification and protonation at the C2 position, ultimately generating an α,β-unsaturated ketone that serves as a Giese-type radical acceptor. We selected 2-azaallyl radicals to engage in Giese radical addition with the in situ generated α,β-unsaturated ketones, furnishing γ-amino ketones—structural analogues of γ-aminobutyric acid (GABA), the principal inhibitory neurotransmitter in the central nervous system and a core scaffold in numerous pharmaceuticals.6,7 Foundational work8 by Walsh and co-workers demonstrated that 2-azaallyl radicals can be formed from N-benzyl ketimines under basic conditions via a single-electron transfer (SET) process between the corresponding 2-azaallyl anion and an electron acceptor (e.g., N-benzyl ketimine). Motivated by our continued interest in α-C–H functionalization of aliphatic amines,9 we sought to develop a Giese-type radical addition between acyl-oxyallenes and N-benzyl ketimines under basic conditions.
Results and discussion
Following these considerations, we selected N-benzyl ketimine 1a (2.0 equiv.) and 3-cyclohexyl-1-phenylprop-2-yn-1-yl benzoate 2a (1.0 equiv.) as model substrates to evaluate the feasibility of the proposed transformation. Under the optimized conditions, the reaction was carried out in 1,4-dioxane at room temperature for 2 hours using KOt-Bu (3.0 equiv.) as the base, affording the desired γ-amino ketone (3aa) in 89% yield (83% isolated) with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio (dr). The effect of various reaction parameters was systematically investigated (Table 1; see the SI for additional data). Modification of the base loading, either increasing or decreasing the amount of KOt-Bu, led to diminished yields (entries 2 and 3). The use of alternative alkoxide bases such as NaOt-Bu and LiOt-Bu also resulted in reduced efficiency (entries 4 and 5). Notably, replacing KOt-Bu with sterically hindered bases such as MHMDS (M = Li, Na, K)—commonly used in 2-azaallyl anion-enabled radical processes8 completely suppressed the reaction (entries 6–8), likely due to steric hindrance impeding the isomerization of the propargylic ester to the corresponding enone intermediate. Further examination of the ester leaving group revealed that propargylic alcohol derivatives bearing alternative activating groups, such as acetyl (Ac) or tert-butylcarbonyl (Boc), provided lower yields (entries 9 and 10). Solvent screening showed that replacing dioxane with toluene or n-hexane significantly decreased the product yield (entries 11 and 12). A reduction in reaction temperature from room temperature to 0 °C also led to a decrease in yield (entry 13). Based on these studies, the optimal conditions were established using KOt-Bu (3.0 equiv.) in dioxane at room temperature for 2 hours (entry 1).
:1 diastereomeric ratio (dr). The effect of various reaction parameters was systematically investigated (Table 1; see the SI for additional data). Modification of the base loading, either increasing or decreasing the amount of KOt-Bu, led to diminished yields (entries 2 and 3). The use of alternative alkoxide bases such as NaOt-Bu and LiOt-Bu also resulted in reduced efficiency (entries 4 and 5). Notably, replacing KOt-Bu with sterically hindered bases such as MHMDS (M = Li, Na, K)—commonly used in 2-azaallyl anion-enabled radical processes8 completely suppressed the reaction (entries 6–8), likely due to steric hindrance impeding the isomerization of the propargylic ester to the corresponding enone intermediate. Further examination of the ester leaving group revealed that propargylic alcohol derivatives bearing alternative activating groups, such as acetyl (Ac) or tert-butylcarbonyl (Boc), provided lower yields (entries 9 and 10). Solvent screening showed that replacing dioxane with toluene or n-hexane significantly decreased the product yield (entries 11 and 12). A reduction in reaction temperature from room temperature to 0 °C also led to a decrease in yield (entry 13). Based on these studies, the optimal conditions were established using KOt-Bu (3.0 equiv.) in dioxane at room temperature for 2 hours (entry 1).
|
|
||
|---|---|---|
| Entrya | Deviation from standard conditions | Yieldb (%) |
|
a Unless otherwise specified, the reactions were performed at rt in dioxane (1 mL) under nitrogen for 2 h with 1a (0.1 mmol), propargyl ester 2a (0.2 mmol) and KOt-Bu (0.3 mmol, 3.0 equiv.).
b Yield was determined by 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard; number in the parentheses refers to isolated yield; the diastereoselectivities (dr, anti/syn ratios) were determined by 1H NMR of the crude reaction mixture, dr = 10:1.
|
||
| 1 | None | 89 (83) |
| 2 | 2.0 equiv. KOt-Bu | 66 |
| 3 | 4.0 equiv. KOt-Bu | 76 |
| 4 | NaOt-Bu vs. KOt-Bu | 20 |
| 5 | LiOt-Bu vs. KOt-Bu | 0 |
| 6 | LiHMDS vs. KOt-Bu | 0 |
| 7 | NaHMDS vs. KOt-Bu | 0 |
| 8 | KHMDS vs. KOt-Bu | 0 |
| 9 | Ac vs. Bz | 60 |
| 10 | Boc vs. Bz | 52 |
| 11 | PhMe vs. dioxane | 30 |
| 12 | n-Hexane vs. dioxane | 40 |
| 13 | 0 °C vs. rt | 50 |
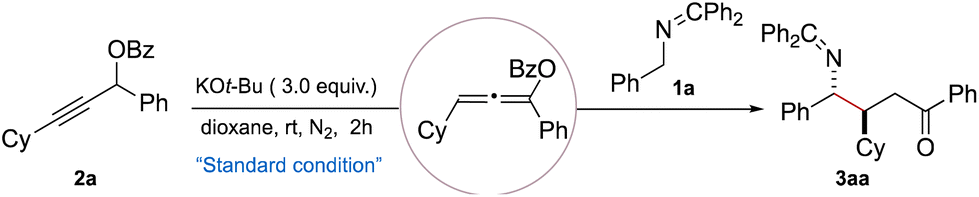
With the optimized conditions in hand, we next explored the substrate scope of the Giese-type addition reaction (Table 2). A wide array of N-benzyl ketimine derivatives (1b–1h) bearing electronically varied substituents, including –Me, –OMe, –SMe, –OPh, –Cl, –Br, and –F at the para-positions of the aromatic ring, were well tolerated, affording the corresponding γ-amino ketones (3ba–3ha) in moderate to excellent yields (60–99%) and with a moderate dr (3:1 to 7:1). Ketimines bearing a chloro substituent at ortho-or meta-positions on the aromatic ring also proved to be suitable substrates, yielding products 3ia and 3ja in 95% and 79% yields respectively, albeit with a lower dr. Moreover, aromatic systems incorporating naphthyl and heteroaryl moieties, such as thiophene and furan, were also compatible with the reaction conditions, furnishing products 3ka–3ma efficiently.
| a Unless otherwise specified, the reactions were performed at rt in dioxane under nitrogen for 2 h with 1 (0.2 mmol), propargyl ester 2 (0.4 mmol) and KOt-Bu (0.6 mmol); the relative configurations were determined or assigned by analogy on the basis of the X-ray structure of 3af; the diastereoselectivities (anti/syn ratios) were determined by 1H NMR of the crude reaction mixture. |
|---|
|
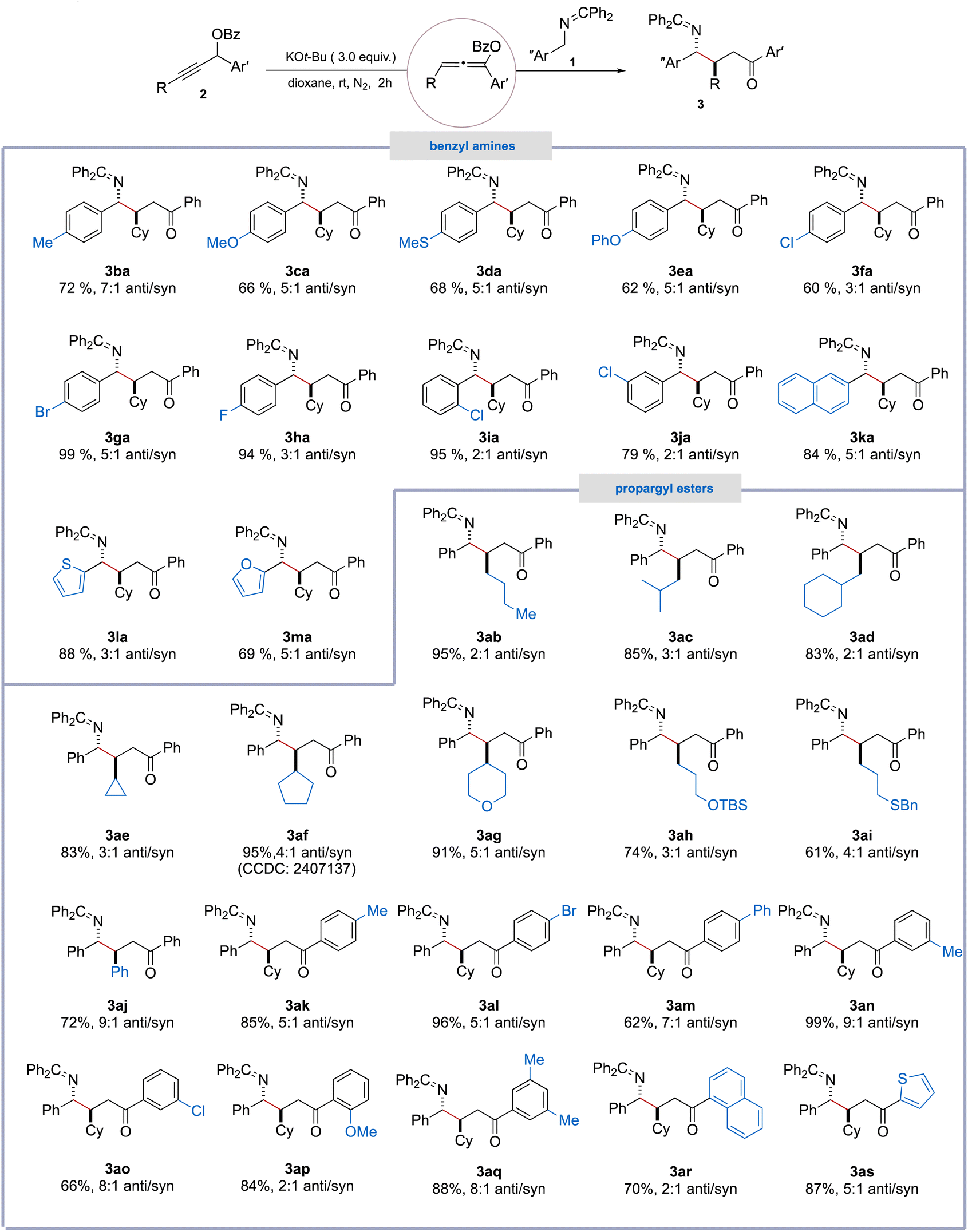
We next turned our attention to evaluating the scope of propargyl ester substrates. A broad range of alkyl-substituted propargylic esters bearing linear, α-branched, β-branched, and γ-branched unfunctionalized alkyl groups on the terminal alkyne carbon, including n-butyl (2b), isobutyl (2c), cyclohexylmethyl (2d), cyclopropyl (2e), and cyclopentyl (2f) groups, underwent smooth reactions with N-benzyl ketimine (1a), affording the corresponding γ-amino ketones (3ab–3af) in high to excellent yields (83–95%), with the dr ranging from 2:1 to 4:1.10 In addition, substituents on the terminal alkyne carbon with diverse functional groups, including tetrahydro-2H-pyran (2g), OTBS (2h), thioether (2i), and phenyl (2j), were found to be compatible with these reaction conditions. Moreover, aryl-substituted propargyl esters bearing electron-donating (−Me and –OMe) or electron-withdrawing groups (–Br, –Cl, and –Ph) on the aromatic ring at para-, or meta- or ortho-positions were found to be well tolerated to furnish the corresponding γ-amino ketones (3ak–3ap) in moderate to excellent yields (62–99%) with the dr varied from 2:1 to 9:1. Additionally, substrates featuring more complex aryl groups, such as 3,5-dimethylphenyl, naphthalenyl, and thiophenyl at the C1-position, also proved to be effective, delivering the corresponding products 3aq–3as in good yields.
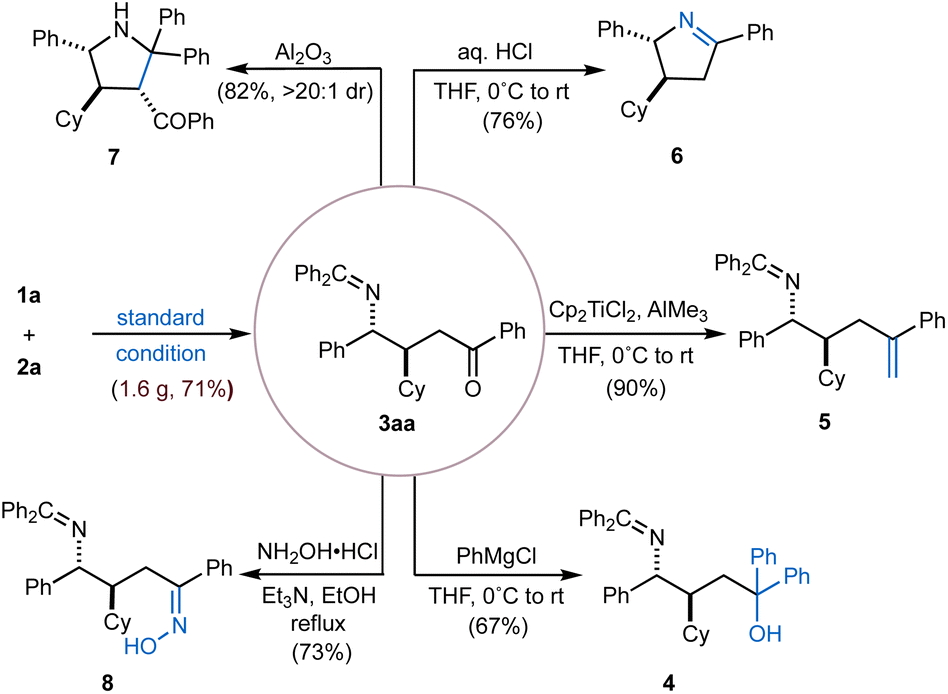
To demonstrate the synthetic utility of this methodology, we performed a gram-scale reaction, which successfully afforded the desired product 3aa in 71% yield. The versatility of this γ-amino ketone scaffold was further highlighted through a series of downstream functionalizations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (Fig. 2). For example, selective nucleophilic addition of PhMgCl furnished the corresponding tertiary alcohol (4) in 67% yield. Alternatively, olefination by employing Tebbe reagent (Cp2TiCl2 and AlMe3) afforded alkene product 5 in 90% yield. Condensation with hydroxylamine hydrochloride delivered oxime 8 in 73% yield. In addition, a cascade sequence involving imine hydrolysis followed by intramolecular condensation under acidic aqueous conditions (aq. HCl), led to the formation of cyclic imine (6) in 76% yield. Finally, intramolecular cyclization of 3aavia a Mannich-type reaction under basic conditions (basic Al2O3) yielded the multi-substituted pyrrolidine (7) in 82% yield with >20:1 dr.
O double bond (Fig. 2). For example, selective nucleophilic addition of PhMgCl furnished the corresponding tertiary alcohol (4) in 67% yield. Alternatively, olefination by employing Tebbe reagent (Cp2TiCl2 and AlMe3) afforded alkene product 5 in 90% yield. Condensation with hydroxylamine hydrochloride delivered oxime 8 in 73% yield. In addition, a cascade sequence involving imine hydrolysis followed by intramolecular condensation under acidic aqueous conditions (aq. HCl), led to the formation of cyclic imine (6) in 76% yield. Finally, intramolecular cyclization of 3aavia a Mannich-type reaction under basic conditions (basic Al2O3) yielded the multi-substituted pyrrolidine (7) in 82% yield with >20:1 dr.
 | ||
| Fig. 2 Synthetic applications. | ||
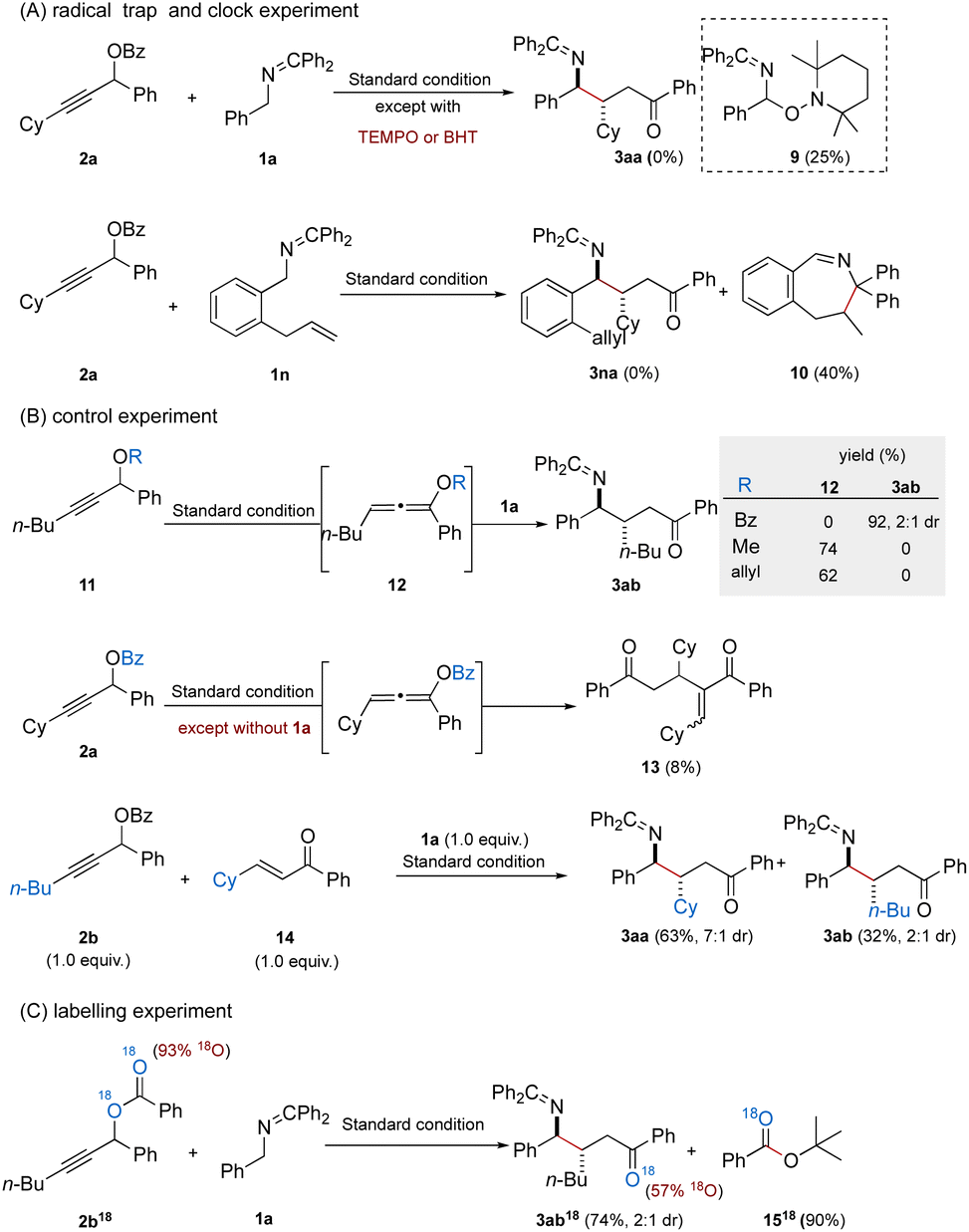
To gain insight into the Giese radical addition, we conducted a series of mechanistic experiments (Fig. 3). First, the addition of radical scavengers such as 2,2,6,6-tetramethylpiperidine N-oxide (TEMPO) or butylated hydroxytoluene (BHT) under standard conditions completely suppressed the formation of the desired γ-amino ketone. Instead, in the presence of TEMPO, a distinct byproduct 9 was isolated in 25% yield, indicating the interception of a 2-azaallyl radical intermediate. Next, the reaction of N-benzyl ketimine (1n), bearing an ortho-allyl substituent on the aromatic ring, with propargyl ester 2a under standard conditions yielded a cyclized product 10 in 40% yield, and no addition product 3na was observed (Fig. 3A). The above observations are consistent with a radical-mediated mechanism, wherein the 2-azaallyl radical could participate in bond forming events at both carbons adjacent to the nitrogen.
 | ||
| Fig. 3 Mechanistic studies. | ||
It was expected that treatment of propargylic benzoate 2a under standard conditions in the absence of ketimine 1a would afford the expected benzoyl-oxyallene or the corresponding enone; however, neither of these intermediates were successfully isolated, only byproduct 13 (8% yield). We reason that both the intermediates are too reactive to be isolated under basic conditions. Benzoyl-oxyallene, under basic conditions (e.g. KOt-Bu), subsequently transformed to enone, which could be consumed by participating in other reaction pathways such as dimerization.11 To illustrate O-acyl oxyallenes as precursors to enones, we performed a series of control experiments (Fig. 3B). When propargylic ether substituted with methyl or allyl groups reacted with N-benzyl ketimine 1a under the standard reaction conditions, the corresponding alkoxyallenes were isolated in moderate yields (74% and 62%) and no formation of the desired product 3ab was observed, likely due to the inert reactivity of these alkoxyallenes, which appear unable to undergo further transformation into the reactive enone intermediate. Next, a competitive experiment was conducted by employing both propargylic benzoate 2b and enone (14) in the presence of 1a under standard conditions. The desired products 3aa and 3ab were obtained in 63% and 32% yield, respectively, thereby supporting the hypothesis that enones serve as the radical acceptors for 2-azaallyl radicals. Finally, an 18O-labeling experiment was performed to elucidate the formation pathway of the enone (Fig. 3C). When 18O-enriched propargylic benzoate 2b18 (93% 18O) was treated with 1a under standard conditions, the resulting product 3ab18 incorporated 57% 18O, and tert-butyl benzoate (1518) was isolated in 90% yield. These findings are consistent with a nucleophilic benzoyl transfer mechanism.
Based on the above experimental observations and the pioneering studies on 2-azaallyl radicals by the Walsh group, we propose a plausible mechanism for this transformation (Fig. 4). The reaction is initiated by the base-mediated isomerization of propargylic benzoate to benzoyl-oxyallene, which undergoes a sequence of benzoyl group transfer and protonation steps at the C2 position of oxyallene, ultimately furnishing the enone in situ. Concurrently, deprotonation of the N-benzyl ketimine by KOt-Bu generates a 2-azaallyl anion, which serves as a super-electron donor. A single-electron transfer (SET) from this anion to another molecule of N-benzyl ketimine produces a 2-azaallyl radical along with a ketimine radical anion.8a The resulting 2-azaallyl radical then undergoes a Giese-type radical addition to the in situ generated enone, forming a new C–C bond and yielding a α-carbonyl radical intermediate. This α-carbonyl radical could react with N-benzyl ketimine via hydrogen atom transfer (HAT) to produce the final product. Alternatively, the SET/PT pathway (SET with either the 2-azaallyl anion or the ketimine radical anion, followed by protonation transfer) is also feasible.
 | ||
| Fig. 4 Plausible mechanism. | ||
Conclusions
In summary, we have revealed that acyl-oxyallenes serve as effective α,β-unsaturated ketone precursors in Giese-type radical additions with 2-azaallyl radicals, culminating in the synthesis of a series of γ-amino ketones. This protocol exhibits remarkable yield, broad substrate scope, and notable functional group tolerance. Moreover, the downstream diversification of the γ-amino ketone products highlights the synthetic utility and pharmaceutical relevance of this methodology. Mechanistic investigations support a radical pathway featuring C–C bond-formation via 2-azaallyl radical addition to transient enone intermediates. These findings expand the synthetic utility of oxyallenes, and efforts to further explore their potential as α,β-unsaturated ketone surrogates are ongoing.Author contributions
J. J., X. L., Y. L., J. C., W. T., and K. D. contributed to the chemical experiments, J. J., X. L., and Y. L. performed all chemical reactions reported, and K. D. and W. T. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2407137 contains the supplementary crystallographic data for this paper.10The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: reaction optimizations, experimental procedures, characterization and X-ray data. See DOI: https://doi.org/10.1039/d5sc08002a.
Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22101234 and 82188101), the National Key R&D Program of China (2022YFA1503702 and 2021YFF0701601), startup funding and the Research Funds of Hangzhou Institute for Advanced Study, UCAS (2024HIAS-Y015 and 2024HIAS-P003).Notes and references
- For selected reviews on alkoxylallenes, see: (a) R. Zimmer, Synthesis, 1993, 165 CrossRef CAS; (b) H.-U. Reissig, S. Hormuth, W. Schade, M. O. Amombo, T. Watanabe, R. Pulz, A. Hausherr and R. Zimmer, J. Heterocycl. Chem., 2000, 37, 597 CrossRef CAS; (c) N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004, p. 425 CrossRef; (d) M. Brasholz, H.-U. Reissig and R. Zimmer, Acc. Chem. Res., 2009, 42, 45 CrossRef CAS PubMed; (e) R. Zimmer and H.-U. Reissig, Chem. Soc. Rev., 2014, 43, 2888 RSC; (f) M. A. Tius, Chem. Soc. Rev., 2014, 43, 2979 RSC; (g) E. Manoni and M. Bandini, Eur. J. Org Chem., 2016, 3135 CrossRef CAS; (h) H.-U. Reissig and R. Zimmer, Synthesis, 2017, 49, 3291 CrossRef CAS; (i) V. M. Schmiedel and H.-U. Reissig, Curr. Org. Chem., 2019, 23, 2976 CrossRef CAS.
- (a) S. Li, J. Lv and S. Luo, Org. Chem. Front., 2018, 5, 1787 RSC; (b) T. Zhang, D. Ji and J. Sun, Org. Biomol. Chem., 2023, 21, 3340 RSC; (c) C. Peng, Q. Guo, G.-X. Xu, L. Huo, W. Wu, T.-Y. Chen, X. Hong and P. Hu, J. Am. Chem. Soc., 2024, 146, 14422 CrossRef CAS; (d) D.-M. Cui, Z.-L. Zheng and C. Zhang, J. Org. Chem., 2009, 74, 1426 CrossRef CAS PubMed; (e) H. Zhou, Z. Wei, J. Zhang, H. Yang, C. Xia and G. Jiang, Angew. Chem., Int. Ed., 2017, 56, 1077 CrossRef CAS PubMed; (f) J. Zhang, L. Zhu, K. Shen, H. Yang, X.-C. Hang and G. Jiang, Chem. Sci., 2019, 10, 1070 RSC.
- For selected recent reviews, see: (a) P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524 CrossRef CAS; (b) G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060 RSC; (c) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545 CrossRef CAS PubMed; (d) H. Wang, Q. Zhang and W. Zi, Acc. Chem. Res., 2024, 57, 468 CrossRef CAS.
- For selected recent reviews, see: (a) M. Xiang, D. E. Pfaffinger and M. J. Krische, Chem.–Eur. J., 2021, 27, 13107 CrossRef CAS PubMed , for selected recent examples, see: ; (b) M. Xiang, D. E. Pfaffinger, E. Ortiz, G. A. Brito and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 8849 CrossRef CAS PubMed; (c) N. Navaneetha, S. Maurya, P. Behera, S. B. Jadhav, L. R. Magham, J. B. Nanubolu, L. Roy and R. Chegondi, Chem. Sci., 2024, 15, 20379 RSC; (d) S. Maurya, N. Navaneetha, P. Behera, J. B. Nanubolu, L. Roy and R. Chegondi, Angew. Chem., Int. Ed., 2025, 64, e202420106 CrossRef CAS.
- For an early example of radical polymerization of alkoxyallenes, see: T. Yokozawa, M. Tanaka and T. Endo, Chem. Lett., 1987, 1831 CrossRef CAS.
- For selected examples of γ-amino acid and derivative synthesis through radical reactions, see: (a) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338 CrossRef CAS PubMed; (b) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef CAS; (c) L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef CAS; (d) A. Millet, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 13464 CrossRef CAS; (e) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS; (f) J. Ma, J. Lin, L. Zhao, K. Harms, M. Marsch, X. Xie and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 11193 CrossRef CAS; (g) X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562 RSC; (h) J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056 CrossRef CAS; (i) A. Trowbridge, D. Reich and M. J. Gaunt, Nature, 2018, 561, 522 CrossRef CAS PubMed; (j) J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037 CrossRef CAS; (k) R. A. Aycock, C. J. Pratt and N. T Jui, ACS Catal., 2018, 8, 9115 CrossRef CAS; (l) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002 CrossRef CAS; (m) Y. Aramaki, N. Imaizumi, M. Hotta, J. Kumagai and T. Ooi, Chem. Sci., 2020, 11, 4305 RSC; (n) L. Leng, Y. Fu, P. Liu and J. M. Ready, J. Am. Chem. Soc., 2020, 142, 11972 CrossRef CAS; (o) S. K. Pagire, N. Kumagai and M. Shibasaki, Chem. Sci., 2020, 11, 5168 RSC; (p) E. Le Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 5357 CrossRef CAS; (q) A. S. K. Lahdenperä, P. D. Bacoş and R. J. Phipps, J. Am. Chem. Soc., 2022, 144, 22451 CrossRef; (r) L.-C. Wang, Y. Yuan, Y. Zhang and X.-F. Wu, Nat. Commun., 2023, 14, 7439 CrossRef PubMed; (s) Y. Qin, R. Cauwenbergh, S. Pradhan, R. Maiti, P. Franck and S. Das, Nat. Commun., 2023, 14, 7604 CrossRef CAS PubMed; (t) Y. Luo, Y. Zhou, F. Xiao, X. He, Z. Zhong, Q.-L. Zhou, W. Cao, X. Liu and X. Feng, ACS Catal., 2024, 14, 12031 CrossRef CAS.
- (a) D. L. Martin and R. W. Olse, GABA in the Nervous System: The View at Fifty Years, Lippincott Williams & Wilkins, 2000 Search PubMed; (b) D.-H. Ngo and T. S. Vo, Molecules, 2019, 24, 2678 CrossRef CAS.
- For selected examples, see: (a) M. Li, O. Gutierrez, S. Berritt, A. Pascual-Escudero, A. Yeşilçimen, X. Yang, J. Adrio, G. Huang, E. Nakamaru-Ogiso, M. C. Kozlowski and P. J. Walsh, Nat. Chem., 2017, 9, 997 CrossRef CAS; (b) M. Li, S. Berritt, L. Matuszewski, G. Deng, A. Pascual-Escudero, G. B. Panetti, M. Poznik, X. Yang, J. J. Chruma and P. J. Walsh, J. Am. Chem. Soc., 2017, 139, 16327 CrossRef CAS PubMed; (c) R. A. Shelp and P. J. Walsh, Angew. Chem., Int. Ed., 2018, 57, 15857 CrossRef CAS; (d) G. Deng, M. Li, K. Yu, C. Liu, Z. Liu, S. Duan, W. Chen, X. Yang, H. Zhang and P. J. Walsh, Angew. Chem., Int. Ed., 2019, 58, 2826 CrossRef CAS; (e) Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2020, 11, 7619 RSC; (f) G. Deng, S. Duan, J. Wang, Z. Chen, T. Liu, W. Chen, H. Zhang, X. Yang and P. J. Walsh, Nat. Commun., 2021, 12, 3860 CrossRef CAS PubMed; (g) D. Zou, L. Gan, F. Yang, H. Wang, Y. Pu, J. Li and P. J. Walsh, Nat. Commun., 2021, 12, 7060 CrossRef CAS PubMed; (h) S. Duan, Y. Zi, L. Wang, J. Cong, W. Chen, M. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2022, 13, 3740 RSC; (i) Y. Jiang, D. Liu, M. E. Rotella, G. Deng, Z. Liu, W. Chen, H. Zhang, M. C. Kozlowski, P. J. Walsh and X. Yang, J. Am. Chem. Soc., 2023, 145, 16045 CrossRef CAS; (j) W.-J. Yue and R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202310304 CrossRef CAS PubMed; (k) Y. Jiang, D. Liu, L. Zhang, C. Qin, H. Li, H. Yang, P. J. Walsh and X. Yang, Chem. Sci., 2024, 15, 2205 RSC.
- Z. Wang, S. Guo, Z. Shen, R. Wei, R. Jiang, G. Liu, Q. Zhou, K. Du and W. Tang, Chem Catal., 2025, 5, 101331 CAS.
- CCDC 2407137: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lstlt.
- F.-Y. Zhang and E. J. Corey, Org. Lett., 2006, 6, 3397 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2026 |