
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible polarization-enabled hydrogen evolution reaction on two-dimensional ferroelectric Cun(CrSe2)n+1 monolayers
Wenyuan
Zhang†
 a,
Jingguo
Wang†
b,
Qi
Wang
a,
Yanling
Si
*b and
Guochun
Yang
*a
a,
Jingguo
Wang†
b,
Qi
Wang
a,
Yanling
Si
*b and
Guochun
Yang
*a
aState Key Laboratory of Metastable Materials Science & Technology, Hebei Key Laboratory of Microstructural Material Physics, School of Science, Yanshan University, Qinhuangdao 066004, China. E-mail: yanggc468@nenu.edu.cn
bSchool of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, China. E-mail: siyl@ysu.edu.cn
First published on 25th February 2026
Abstract
The scalable production of hydrogen through electrochemical water splitting demands earth-abundant catalysts with both high activity and dynamic tunability, yet achieving these attributes simultaneously remains a major challenge. Two-dimensional (2D) ferroelectric materials offer a unique opportunity, as their reversible polarization can modulate surface electronic states, though their potential in electrocatalysis has scarcely been explored. Here, we employ first-principles calculations to investigate the electronic structure and hydrogen evolution reaction (HER) activity of recently synthesized Cun(CrSe2)n+1 (n = 1–3) monolayers with tunable thickness and robust multiferroic behavior at room temperature. We identify surface Se top sites as the optimal catalytic centers, with the down-polarized state exhibiting HER activity comparable to that of benchmark Pt(111). A strong inverse correlation between hydrogen adsorption free energy and the p-band center of surface Se atoms is further established, providing a predictive descriptor for catalyst design. Crucially, reversible polarization dynamically modulates hydrogen adsorption energetics through charge redistribution, enabling efficient transitions between H adsorption and H2 desorption and thereby maximizing HER efficiency. These insights position Cun(CrSe2)n+1 as a promising polarization-switchable platform for high-performance and controllable electrocatalysis, offering general design principles for next-generation ferroelectric catalysts.
1 Introduction
The global energy crisis and environmental pollution pose significant challenges to sustainable development.1–3 Hydrogen, as a clean and renewable energy carrier, has garnered tremendous interest due to its abundance, high gravimetric energy density, and zero carbon emissions.4–6 Electrochemical water splitting has emerged as a promising and scalable technology for sustainable hydrogen production, owing to its inherent safety, modularity, and environmental compatibility.7–9 However, the efficiency of this process is fundamentally limited by the sluggish kinetics of the hydrogen evolution reaction (HER), which necessitates the use of highly active and durable electrocatalysts.10,11 Platinum (Pt) remains the state-of-the-art catalyst for the HER, thanks to its near-optimal hydrogen adsorption energy, which provides crucial support for its exceptional catalytic activity.12–14 Yet, the scarcity and exorbitant cost of Pt severely restrict its widespread application,15,16 representing a critical bottleneck for the large-scale deployment of water electrolysis in a future hydrogen economy.17,18Addressing this challenge requires a paradigm shift from simply replacing Pt with cheaper alternatives to fundamentally rethinking catalyst design. Emerging strategies, including electronic structure modulation,19–21 interfacial engineering,22–24 and defect manipulation,25–27 have shown great promise in tuning adsorption energetics, accelerating charge transfer, and enhancing catalytic stability under operational conditions. In particular, the exploration of unconventional active sites,28 such as low-coordination metal centers,29 heteroatom-doped frameworks,30 and atomically dispersed catalysts,31 has expanded the design landscape, breaking traditional activity–stability trade-offs. Despite these advances, realizing practical, scalable electrocatalysts that combine intrinsic activity, robustness, and manufacturability remains a formidable challenge, underscoring the need for predictive, theory-guided frameworks that seamlessly integrate atomic-scale design with experimental validation.
Two-dimensional (2D) materials have attracted considerable attention as versatile electrocatalytic platforms due to their high surface-to-volume ratio, abundant active sites, and tunable electronic properties.32,33 Among them, 2D ferroelectric materials offer unique mechanistic advantages for catalysis because their switchable polarization can dynamically modulate the local electronic environment at surface sites.34–37 This polarization-induced charge redistribution directly affects the binding strength of reaction intermediates, enabling the breaking of conventional linear scaling relationships and precise control over reaction energetics.37–39 Experimental and theoretical studies have demonstrated that polarization reversal can systematically tune intermediate binding energies, break linear scaling relationships, and enhance HER activity in diverse systems such as BaTiO3,40 CuInP2S6,36 L-Bi2O2Se,41 CuCrS2 (ref. 42) and TMN3/In2Se3 heterostructures.43 These observations establish ferroelectric polarization as a mechanistic lever for achieving controllable and tunable catalytic performance beyond the limitations of conventional electrocatalysts.
Motivated by these insights, it is essential to identify intrinsically stable 2D ferroelectrics capable of delivering polarization-tunable HER performance. Recently, 2D ferroelectric CuCrSe2 monolayers have been synthesized through chemical exfoliation under redox control as well as through chemical vapor deposition.44–46 To avoid confusion, we refer to the exfoliated layered materials as Cun(CrSe2)n+1 (n = 1–3), where n denotes the number of layers. Experimental and theoretical studies have reported high phase-transition temperatures and multiferroic behavior in these systems.44,46,47 Their adjustable thickness and inherent ferroelectricity make them attractive candidates for electrocatalytic hydrogen evolution. Here, we employ density functional theory to systematically explore the dynamical stability, electronic structure, and HER activity of Cun(CrSe2)n+1 (n = 1–3). We identify the top Se site as the optimal active center and find that all structures in the down-polarized state show superior HER activity. A strong inverse correlation between hydrogen adsorption free energy and the Se p-band center is revealed, establishing it as a reliable descriptor. Crucially, polarization reversal dynamically adjusts hydrogen adsorption energetics through charge redistribution, enabling hydrogen adsorption and desorption to proceed via distinct electronic states and thereby markedly accelerating the entire HER process. These results position Cun(CrSe2)n+1 (n = 1–3) as a promising platform for polarization-tunable HER electrocatalysis.
2 Calculation methods
All spin-polarized first-principles calculations are performed based on the density functional theory (DFT) using the Vienna ab initio Simulation Package (VASP).48–50 The exchange–correlation interactions are treated using the Perdew–Burke–Ernzerhof (PBE) functional within the framework of the Generalized Gradient Approximation (GGA),49,51 and DFT-D3 dispersion correction is applied to account for van der Waals interactions. The DFT + U method (UCr = 3 eV)52 is employed to account for the strong correlation of the 3d electrons of Cr.53 The plane wave cutoff energy is set to 520 eV. A vacuum layer of 15 Å is added in the z-direction to prevent interactions between adjacent layers, and dipole corrections are included to eliminate artifacts from periodic boundary conditions. A 2 × 2 × 1 supercell is used to investigate the hydrogen evolution catalytic performance of Cun(CrSe2)n+1 (n = 1–3). The Brillouin zone is sampled using 2π × 0.03 Å−1. All models undergo structural relaxation until the force convergence and energy convergence are within 0.03 eV Å−1 and 10−5 eV, respectively. The PBE functional is employed to analyze the band structure and density of states. The lattice dynamical stability and phonon dispersion curves are computed using the supercell method implemented in the Phonopy code.54 The thermal stability is achieved through ab initio molecular dynamics simulations (AIMD) using the Nosé-Hoover heat bath method under an NVT ensemble at 400 K for 5 ps.55 The hydrogen evolution reaction barrier is calculated using the climbing image nudged elastic band (CI-NEB) method implemented in the VASP.The formulas for calculating the in-plane Young's modulus and Poisson's ratio at any angle are as follows:56
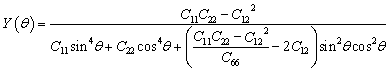 | (1) |
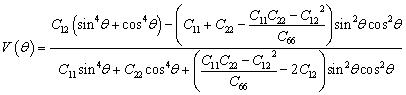 | (2) |
The hydrogen evolution reaction performance of Cun(CrSe2)n+1 (n = 1–3) is evaluated by calculating ΔGH*, which can be computed using the following formula:
| ΔGH* = ΔEH + ΔEZPE − TΔSH | (3) |
In this context, ΔEH, ΔEZPE and ΔSH represent the adsorption energy of H atom, the zero-point energy difference between adsorbed hydrogen and gaseous hydrogen, and the entropy difference, respectively. T denotes room temperature, and the value of ΔEZPE − TΔSH is 0.24 eV.43,57 The adsorption energy of hydrogen can be calculated using the following equation:
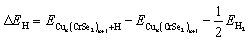 | (4) |
Among them, ECun(CrSe2)n+1, ECun(CrSe2)n+1+H and EH2 are the energies of Cun(CrSe2)n+1 (n = 1–3) before and after H atom adsorption and the total energy of the isolated H2 gas molecule, respectively. Additionally, the exchange current density can be calculated using the following equation:58,59
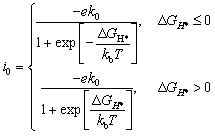 | (5) |
3 Results and discussion
3.1 Structural stability, electronic properties, and ferroelectric behavior
The optimized atomic configurations of Cun(CrSe2)n+1 (n = 1–3, space group P3m1) are illustrated in Fig. 1a, b and S1. They consist of alternating layers of 1T-CrSe2 and Cu, with a lattice constant of 3.58 Å, close to the experimental value46 and previous theoretical ones.53 The stability and polarization of catalysts are crucial for electrocatalytic hydrogen evolution. Building on the experimental synthesis of CuCrSe2-based ferroelectrics, we further investigated the microscopic stability and origin of ferroelectricity in Cun(CrSe2)n+1. Our phonon dispersion calculations (Fig. S2) show no significant imaginary frequencies, indicating dynamical stability. Furthermore, AIMD simulations performed at 400 K, a temperature relevant to practical water electrolysis, reveal only minor energy fluctuations with preserved structural integrity throughout the 5 ps trajectory (Fig. S3), confirming robust thermal stability under operating conditions. To assess mechanical stability, we calculated elastic constants using the energy-strain method (Table S1). All systems satisfy the Born–Huang criteria60 (C11 > 0, C66 > 0, and C11 > |C12|), confirming their mechanical stability. The in-plane Young's modulus and Poisson's ratio (Fig. 1c, d, and S4) are isotropic and the values of both parameters increase with layer number. The minimum Young's modulus of Cu(CrSe2)2 (57.75 N m−1) is comparable to that of α-In2Se3 (53.98 N m−1),61 while the maximum value for Cu3(CrSe2)4 (172.07 N m−1) approaches that of MoS2 (180 N m−1).62 The Poisson's ratios of Cu(CrSe2)2, Cu2(CrSe2)3, and Cu3(CrSe2)4 are 0.23, 0.25, and 0.27, respectively, comparable to those of CrS2 (0.24)63 and CrSO (0.28).64 These results indicate that Cun(CrSe2)n+1 (n = 1–3) possesses tunable mechanical flexibility, controlled by the number of layers. | ||
| Fig. 1 (a and b) Top and side views of the Cu(CrSe2)2 structure, with dashed lines outlining its unit cell. (c) Polar plots showing (c) the Young's modulus and (d) the Poisson's ratio for the Cu(CrSe2)2 structure. (e) Electronic band structure of Cu(CrSe2)2. (f) Projected density of states (PDOS) of Cu(CrSe2)2. | ||
For high-performance electrochemical catalysts, excellent electrical conductivity is essential, and intrinsic metallicity is particularly desirable. The electronic band structure and PDOS of Cun(CrSe2)n+1 (n = 1–3) are shown in Fig. 1e, f, and S5. All three structures exhibit multiple bands crossing the Fermi level, confirming metallic behavior, which primarily arises from Se 4p orbitals. Below the Fermi level, pronounced overlap between Cu/Cr d orbitals and Se p orbitals indicates strong interactions between Cu, Cr, and Se that are responsible for structural stability. As shown in Fig. 1b, the asymmetric vertical distances between Cu and the adjacent Se atoms above and below generate an upward ferroelectric polarization (P↑), denoted as U-state. Under an applied external electric field, Cu atoms undergo both longitudinal and lateral displacements, reversing the polarization to a downward state (P↓), denoted as D-state. This polarization reversal substantially redistributes charge density, offering a dynamic route to tune the electrocatalytic activity of Cun(CrSe2)n+1 for hydrogen evolution.
3.2 Ferroelectric hydrogen evolution activity
According to previous studies, the Gibbs free energy of hydrogen adsorption (ΔGH*) is a widely accepted descriptor for evaluating catalytic performance.65,66 Following the Sabatier principle, the closer ΔGH* is to 0 eV, the more favorable the electrocatalytic activity.46,67–69 This means that an ideal HER electrocatalyst should bind hydrogen neither too strongly nor too weakly, ensuring efficient adsorption and desorption. Specifically, |ΔGH*| ≤ 0.20 eV is generally regarded as a criterion for high-performance HER catalysts.65 To identify the active sites, we employed a 2 × 2 × 1 supercell for Cu(CrSe2)2, Cu2(CrSe2)3, and Cu3(CrSe2)4, considering high-symmetry adsorption sites based on geometric symmetry (Fig. 1a). These sites include the top sites of Se atoms and Cr atoms (TSe and TCr) and the hollow sites of the wrinkled hexagonal rings formed by Se and Cr atoms (HSeCr). Using eqn (3), we calculated ΔGH* at these sites under different polarization states (Table S2) and constructed HER free-energy diagrams (Fig. 2a–c). | ||
| Fig. 2 (a–c) Gibbs free energy profile of the hydrogen evolution reaction for Cun(CrSe2)n+1 (n = 1–3). The hydrogen evolution reaction mechanism on the top surface of Cu(CrSe2)2 in the downward polarization (D) state: (d) Volmer process, (e) Heyrovsky process, and (f) Tafel process, along with schematic diagrams of the corresponding reaction structures. | ||
The results demonstrate that in the D-state, the top-layer Se site (TSe) consistently provides the most favorable hydrogen adsorption in all Cun(CrSe2)n+1 (n = 1–3) systems. The ΔGH*values at TSe(D) are −0.07 eV, −0.10 eV, and −0.14 eV for Cu(CrSe2)2, Cu2(CrSe2)3, and Cu3(CrSe2)4 respectively, meeting the high-performance criterion. TCr(D) sites in Cu(CrSe2)2 and Cu3(CrSe2)4 also exhibit reasonable activity (|ΔGH*| = 0.10 eV and 0.11 eV), while Cu2(CrSe2)3 shows weaker performance (ΔGH* = 0.21 eV). Hollow sites support H adsorption only in Cu2(CrSe2)3 in the D-state, but with a large ΔGH* of 0.91 eV, indicating HER inertness. In contrast, all sites in the U-state display poor HER performance. Interestingly, electronic-structure analysis confirms that Cun(CrSe2)n+1 (n = 1–3) preserve metallicity after hydrogen adsorption (Fig. S6a), with Cr and Cu d states contributing to the metallic character and ensuring efficient electron transfer during the HER process.
Building on dispersion and dipole corrections, we incorporated implicit solvent effects using VASPsol (ε = 78.4)70 to recalculate ΔGH* at the TSe(D) site of Cu(CrSe2)2. As shown in Fig. S7, solvation has a minimal effect on the energetics and structure: ΔGH* shifts by only 0.09 eV (from −0.07 to 0.02 eV), while the Se–H bond length changes negligibly from 1.484 to 1.485 Å. These results indicate that solvation effects are minor, justifying their omission in subsequent calculations and discussions to reduce computational cost.
Focusing on Cu(CrSe2)2, the HER proceeds via two fundamental steps: hydrogen adsorption (Volmer step), followed by hydrogen desorption through either the Heyrovsky or the Tafel pathway. In acidic aqueous electrolytes, protons exist predominantly in the form of hydronium ions (H3O+), which diffuse to the electrode surface and participate in interfacial charge-transfer processes.71 The Volmer reaction (H3O+ + e− → H* + H2O) involves the reduction of hydronium and the formation of adsorbed hydrogen species (H*) on the catalyst surface. Subsequent hydrogen desorption can occur via the Heyrovsky reaction (H* + e− + H3O+ → H2 + H2O), where an adsorbed hydrogen atom reacts with a solvated proton–electron pair, or via the Tafel reaction (H* + H* → H2), where two adjacent H* species recombine directly to form molecular hydrogen. Among these elementary steps, the one associated with the highest energy barrier governs the overall reaction kinetics. To more realistically describe proton transfer in an aqueous electrochemical environment, we constructed an explicit hydrated proton model based on the Eigen cation (H9O4+), consisting of a central H3O+ core coordinately stabilized by three surrounding H2O molecules.72,73 Bader charge analysis indicates that the H9O4+ cluster carries a net positive charge of +0.62,74 implying partial electron transfer from the hydrated proton complex to the catalyst surface during the Volmer process. As shown in the calculated reaction profile for Cu(CrSe2)2 (Fig. 2d), hydrogen adsorption via the Volmer step is thermodynamically favorable, exhibiting a mildly exothermic adsorption energy of −0.09 eV and an ultra-low activation barrier of ∼0.09 eV. This value is comparable to reported Volmer barriers for representative transition-metal dichalcogenide catalysts, such as the Volmer step on 1T-MoS2 modeled with an explicit solvent model,75 and is significantly lower than the 0.67 eV barrier on the Zn surface under similar theoretical treatments.76 In comparison, the subsequent hydrogen evolution steps are associated with appreciable kinetic barriers (Fig. 2e and f). The activation energy of the Heyrovsky step is calculated to be 0.71 eV, slightly higher than the reported value of 0.57 eV for Pt,77 while the Tafel step exhibits a substantially larger barrier of 1.29 eV. The prohibitively high barrier of the Tafel reaction renders this pathway kinetically unfavorable, suggesting that the HER on Cu(CrSe2)2 predominantly proceeds through a Volmer–Heyrovsky mechanism.
The reversal of ferroelectric polarization markedly alters the charge distribution within the structure, thereby modulating hydrogen adsorption. Fig. 3a–c present the PDOS for the Se-4p and H-1s orbitals in three representative compounds under opposite polarization states. For Cu2(CrSe2)3 in the U-state, the overlap between Se-4p and H-1s orbitals below the Fermi level is relatively small, indicating weak Se–H interactions. Upon reversing the D-state, this orbital overlap increases markedly, reflecting stronger Se–H coupling. Consistently, the electrostatic potential of the surface Se atom is higher in the D-state than in the U-state (Fig. S6b), favoring electron transfer from H and thus strengthening hydrogen adsorption. Bader charge analysis further supports this trend, showing a larger charge transfer from H to Se in the D state (0.009 e) compared with the U state (0.008 e). Thus, ΔEH* = −0.31 eV in the D state is more favorable for hydrogen adsorption than 0.12 eV in the U state. Similar polarization-dependent behavior is observed in Cu2(CrSe2)3 and Cu3(CrSe2)4, demonstrating that switching ferroelectric polarization enables reversible and tunable control of catalytic hydrogen adsorption.
 | ||
| Fig. 3 Projected density of states (PDOS) of Se and H on the upper surface of Cun(CrSe2)n+1 (n = 1–3) in both (D) and (U) spin-polarized states (a–c), together with the average p-band center (εp) of the corresponding surface Se atoms (d–f). For clarity, εp is indicated by a solid line, and the Fermi energy level by a dashed line. | ||
The electronic structure of catalyst surfaces plays a pivotal role in regulating catalytic activity.78 The d-band center theory has been widely used to correlate the binding strength of adsorbates with the electronic states of metal sites.79–82 By analogy, the p-band center theory provides a descriptor for the catalytic activity of nonmetal atoms.83,84 Based on this framework, we computed the average p-band center (εp) of surface Se atoms in Cu(CrSe2)2, Cu2(CrSe2)3, and Cu3(CrSe2)4 under different polarization states. As shown in Fig. 3d–f, the εp values in the D state are −0.72 eV, −0.77 eV, and −0.82 eV, respectively, whereas in the U state they shift to −1.04 eV, −1.11 eV, and −1.17 eV, respectively. These shifts highlight the strong dependence of εp on polarization reversal. According to the p-band center theory, Se atoms with εp closer to the Fermi level exhibit stronger HER activity, consistent with our calculated |ΔGH*| values. To quantify this correlation, we fitted |ΔGH*| against εp for Cun(CrSe2)n+1 (n = 1–3), as shown in Fig. 4a. A clear linear relationship (R2 = 0.97) was obtained, revealing a negative correlation: as εp approaches the Fermi level, |ΔGH*| tends toward zero. This establishes εp of surface Se atoms as a reliable descriptor for the HER activity of these compounds. Finally, the exchange current density (i0) was calculated (eqn (5)) and plotted in a volcano diagram (Fig. 4b). Remarkably, the TSe sites of Cu(CrSe2)2, Cu2(CrSe2)3, and Cu3(CrSe2)4 in the D-state all lie at the apex of the volcano plot, comparable to the benchmark Pt(111), while in the U-state, they all deviate from the top of the volcano diagram. These findings underscore that ferroelectric polarization switching affords effective and reversible regulation of HER performance in Cun(CrSe2)n+1 catalysts.
 | ||
| Fig. 4 Relationship between the hydrogen evolution reaction |ΔGH*| and εp in downward (D) and upward (U) polarization states for Cun(CrSe2)n+1 (n = 1–3) (a), along with the corresponding volcano plot of the i0 (b). Illustration of the potential mechanism by which alternating polarization regulates and accelerates the hydrogen evolution reaction of Cu(CrSe2)2 during electrocatalysis (c). | ||
3.3 Polarization-driven mechanism for the HER
To elucidate the intrinsic mechanism of the polarization-tunable electrocatalytic HER in the Cun(CrSe2)n+1 (n = 1–3) series, Cu(CrSe2)2 is selected as a representative model for detailed analysis. Ferroelectric polarization serves as an effective switch that regulates surface catalytic activity by modulating the surface charge distribution, thereby directly governing the hydrogen adsorption–desorption process. As illustrated Fig. 4c, in the D-state, electronic charge mainly accumulates in the p orbitals of the surface Se active sites. This localized charge enrichment favors hydrogen adsorption, yielding an adsorption energy of −0.31 eV and a corresponding ΔGH* of −0.07 eV. Upon polarization switching to the U-state, a directional interlayer charge redistribution occurs, with electrons transferring from the top CrSe2 layer to the bottom CrSe2 layer and becoming concentrated in the p orbitals of the bottom Se atoms. Consequently, the surface Se sites experience a pronounced depletion of charge density, which weakens hydrogen binding, resulting in a positive adsorption energy of 0.12 eV and thereby facilitating hydrogen desorption. By reversing the polarization direction through an external electric field, the adsorption and desorption steps can be selectively optimized. In contrast to conventional HER catalysts that operate with a fixed surface electronic structure, this polarization-tuning strategy dynamically decouples the optimal electronic states for hydrogen adsorption and desorption, effectively lowering the H2 desorption barrier and accelerating the overall reaction kinetics. Such polarization-driven control provides an efficient route to enhance HER performance.We further assess the ferroelectric integrity and structural stability of Cu(CrSe2)2 under polarization-driven catalytic conditions. Intrinsic ferroelectric polarization in Cu(CrSe2)2 has been experimentally established, with reported coercive fields in the range of 0.25–0.32 V Å−1.44 Consistent with these experimental observations, our calculations yield an intrinsic polarization switching barrier of 0.43 eV, comparable to the previously reported value of 0.33 eV.47 Upon hydrogen adsorption, the switching barrier is moderately reduced to 0.32 eV, indicating that ferroelectric polarization reversal remains energetically accessible under catalytic conditions (Fig. S8). To further examine field-driven switching, external electric fields ranging from −0.5 to +0.5 V Å−1, exceeding the experimentally measured coercive field, are applied in our simulations. Notably, both the lattice parameters and key bonding distances exhibit only negligible variations, irrespective of hydrogen adsorption (Tables S3 and S4), demonstrating robust structural stability under polarization switching. Moreover, when the adsorbed hydrogen atoms are removed and ab initio molecular dynamics simulations are repeated (Fig. S9), the slightly distorted Cu(CrSe2)2 structure rapidly relaxes back to its initial configuration. Collectively, these results suggest that Cu(CrSe2)2 could retain ferroelectric polarization switching while preserving structural integrity under polarization-driven HER conditions, highlighting its potential as a ferroelectrically tunable electrocatalyst.
From a broader perspective, dynamic modulation strategies, such as cyclic polarization switching,38,39,85,86 have been explored to temporally decouple adsorption and desorption processes in the HER, thereby tuning the surface intermediate energetics.40,87,88 In this framework, ferroelectric polarization provides a physically transparent handle to reversibly reshape the surface electronic structure and intermediate binding energetics. For Cu(CrSe2)2, switching from the D-state to the U-state leads to a pronounced reduction in hydrogen desorption barriers (Fig. S10): the Heyrovsky barrier decreases from 0.71 eV to 0.36 eV, while the Tafel barrier decreases from 1.29 eV to 0.72 eV. The corresponding barrier reductions (0.35 eV for Heyrovsky and 0.57 eV for Tafel) both exceed the calculated ferroelectric switching barrier of Cu(CrSe2)2 (0.32 eV). At the elementary-step level, these barrier reductions indicate that the kinetic gain from polarization switching could offset the energetic cost, and its benefit is expected to dominate under moderate switching frequencies.
4 Conclusion
In conclusion, our density functional theory study reveals that two-dimensional ferroelectric Cun(CrSe2)n+1 (n = 1–3) monolayers combine excellent structural stability with outstanding HER activity. Surface Se atoms are identified as optimal active sites, with all structures in the down-polarized state exhibiting hydrogen adsorption energetics comparable to those of benchmark Pt(111). A robust inverse correlation between the hydrogen adsorption free energy and the p-band center of surface Se atoms is established, providing a reliable and physically transparent descriptor for catalytic performance. Importantly, these materials possess reversible ferroelectric polarization, enabling polarization-defined redistribution of surface charge density and access to distinct electronic states that separately favor hydrogen adsorption or desorption. Such state-selective tunability distinguishes ferroelectric catalysts from conventional static electrocatalysts and offers a new perspective on regulating reaction energetics at the atomic scale. The combination of high intrinsic activity, metallic conductivity, and field-addressable polarization responsiveness highlights Cun(CrSe2)n+1 as a promising platform for polarization-tunable electrocatalysis. While the present study focuses on this specific system, the results suggest that ferroelectric polarization may generally provide a means to modulate surface electronic states and influence adsorption–desorption energetics in other catalytic materials, offering guidance for the exploration of polarization-responsive electrocatalysts in future studies.Author contributions
Wenyuan Zhang: conceptualization, writing – original draft, visualization, data curation. Jingguo Wang: conceptualization, writing – original draft, visualization, data curation. Qi Wang: validation, visualization, investigation. Yanling Si: writing – review and editing, project administration, supervision. Guochun Yang: writing – review and editing, conceptualization, funding acquisition, project administration, supervision, formal analysis.Conflicts of interest
The authors declare no competing interests.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Additional data are available from the authors upon reasonable request. Supplementary information: the adsorption energy (ΔEH) and Gibbs free energy (ΔGH*) of hydrogen at the adsorption sites on Cun(CrSe2)n+1 (n = 1–3) surfaces; mechanical property parameters; lattice parameters, phonon spectra of the Cun(CrSe2)n+1 monolayer, and the AIMD simulation at 300 K; structural diagrams, Young's modulus and Poisson's ratio, band structure and density of states of Cu2(CrSe2)3 and Cu3(CrSe2)4; ferroelectric polarization inversion barrier of Cu(CrSe2)2. See DOI: https://doi.org/10.1039/d5sc07802d.Acknowledgements
This work was supported by the National Natural Science Foundation of China under grant no. 22372142, the Foreign Expert Introduction Program (G2023003004L), the Central Guiding Local Science and Technology Development Fund Projects (236Z7605 G), the Natural Science Foundation of Hebei Province (grant no. B2024203051, A2024203023, and A2024203002), the Science and Technology Project of Hebei Education Department (grant no. JZX2023020), Innovation Capability Improvement Project of Hebei province (22567605H), and the Hebei Province Yan Zhao Huang Jin Tai Talent Program (Postdoctoral Platform, B2024003003).References
- M. Elimelech and W. A. Phillip, Science, 2011, 333, 712–717 Search PubMed.
- Y. Guan, J. Yan, Y. Shan, Y. Zhou, Y. Hang, R. Li, Y. Liu, B. Liu, Q. Nie, B. Bruckner, K. Feng and K. Hubacek, Nat. Energy, 2023, 8, 304 Search PubMed.
- Z. Yu, Y. Su, R. Gu, W. Wu, Y. Li and S. Cheng, Nano-Micro Lett., 2023, 15, 214 Search PubMed.
- H.-M. Zhang, S.-F. Zhang, L.-H. Zuo, J.-K. Li, J.-X. Guo, P. Wang, J.-F. Sun and L. Dai, Rare Met., 2024, 43, 2371 CrossRef CAS.
- J. Yang, T. Y. Lam, Z. Luo, Q. Cheng, G. Wang and H. Yao, Renewable Sustainable Energy Rev., 2025, 218, 115804 Search PubMed.
- Z.-Y. Yu, Y. Duan, X.-Y. Feng, X. Yu, M.-R. Gao and S.-H. Yu, Adv. Mater., 2021, 33, 2007100 Search PubMed.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 Search PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148 RSC.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550 CrossRef CAS PubMed.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. Mørch Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175 CrossRef CAS PubMed.
- J. Cui, X. Yu, X. Li, J. Yu, L. Peng and Z. Wei, Chin. J. Catal., 2025, 69, 17–34 CrossRef.
- Y. Song, Y. Zhang, W. Gao, C. Yu, J. Xing, K. Liu and D. Ma, Chem. Sci., 2024, 15, 9851–9857 RSC.
- S. Wu, Z. Wang, H. Zhang, J. Cai and J. Li, Energy Environ. Mater., 2023, 6, e12259 CrossRef CAS.
- X. Tian, X. Zhao, Y.-Q. Su, L. Wang, H. Wang, D. Dang, B. Chi, H. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Science, 2019, 366, 850 CrossRef CAS PubMed.
- X. Sun, P. Zhang, B. Zhang and C. Xu, Small, 2024, 20, 2405624 Search PubMed.
- R. Cheng, Y. Min, H. Li and C. Fu, Nano Energy, 2023, 115, 108718 CrossRef CAS.
- H. Li, H. Li, M. Du, E. Zhou, W. R. Leow and M. Liu, Chem. Sci., 2025, 16, 1506–1527 RSC.
- B. Xu, J. Liang, X. Sun and X. Xiong, Green Chem., 2023, 25, 3767 RSC.
- Y. Zhang, Y. Lin, T. Duan and L. Song, Mater. Today, 2021, 48, 115 CrossRef CAS.
- J. Liu, P. Li, S. Jia, Y. Wang, L. Jing, Z. Liu, J. Zhang, Q. Qian, X. Kang, X. Sun, Q. Zhu and B. Han, Nat. Synth., 2025, 4, 730–743 CrossRef CAS.
- Y. Lu, L. Zhou, S. Wang and Y. Zou, Nano Res., 2023, 16, 1890 CrossRef CAS.
- H.-J. Niu, N. Ran, W. Zhou, W. An, C. Huang, W. Chen, M. Zhou, W.-F. Lin, J. Liu and L. Guo, J. Am. Chem. Soc., 2025, 147, 2607 CrossRef CAS PubMed.
- W. Ma, J. Yao, F. Xie, X. Wang, H. Wan, X. Shen, L. Zhang, M. Jiao and Z. Zhou, Green Energy Environ., 2025, 10, 109 CrossRef CAS.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- L. Yuan, S. Zeng, G. Li, Y. Wang, K. Peng, J. Feng, X. Zhang and S. Zhang, Adv. Funct. Mater., 2023, 33, 2306994 CrossRef CAS.
- J. Wang, Y. Zhu, N. Wu, Y. Kan and Y. Hu, Composites, Part B, 2022, 233, 109644 Search PubMed.
- X. Cai, X. Chen, Z. Ying, S. Wang, Y. Chen, Y. Cai, G. Long, H. Liu and N. Wang, Mater. Des., 2021, 210, 110080 CrossRef CAS.
- X. Hu, S. Zhang, J. Sun, L. Yu, X. Qian, R. Hu, Y. Wang, H. Zhao and J. Zhu, Nano Energy, 2019, 56, 109 CrossRef CAS.
- Z. Cui, R. Sa, W. Du, C. Xiao, Q. Li and Z. Ma, Appl. Surf. Sci., 2021, 542, 148535 CrossRef CAS.
- M. Li and J. Young, J. Mater. Chem. A, 2024, 12, 10252–10259 RSC.
- X. Li, Y. Du, L. Ge, C. Hao, Y. Bai, Z. Fu, Y. Lu and Z. Cheng, Adv. Funct. Mater., 2023, 33, 2210194 CrossRef CAS.
- F. Wang, L. Ju, B. Wu, S. Li, J. Peng, Y. Chen, M. Getaye Sendeku, K. Wang, Y. Cai, J. Yi, Y. Yang, Z. Wang and X. Sun, Angew. Chem., Int. Ed., 2024, 63, e202402033 CrossRef CAS PubMed.
- H. S. Kim, J. Mater. Chem. A, 2021, 9, 11553–11562 RSC.
- A. Kakekhani and S. Ismail-Beigi, ACS Catal., 2015, 5, 4537 CrossRef CAS.
- L. Ju, X. Tan, X. Mao, Y. Gu, S. Smith, A. Du, Z. Chen, C. Chen and L. Kou, Nat. Commun., 2021, 12, 5128 CrossRef CAS PubMed.
- P. Abbasi, M. R. Barone, M. de la Paz Cruz-Jáuregui, D. Valdespino-Padilla, H. Paik, T. Kim, L. Kornblum, D. G. Schlom, T. A. Pascal and D. P. Fenning, Nano Lett., 2022, 22, 4276 CrossRef CAS PubMed.
- C.-H. Chiang, C.-H. Yu, Y.-S. Lu, Y.-C. Yang, Y.-C. Lin, H.-A. Chen, S.-Z. Ho, Y.-C. Chen, A. Kumatani, C. Chang, P.-C. Kuo, J. Shiue, S.-S. Li, P.-W. Chiu and C.-W. Chen, Nano Lett., 2024, 24, 11012–11019 CrossRef CAS PubMed.
- Y. Wang, C. Lu and M. Wu, Comput. Mater. Today, 2025, 6, 100029 CrossRef.
- T. L. Wan, J. Liu, X. Tan, T. Liao, Y. Gu, A. Du, S. Smith and L. Kou, J. Mater. Chem. A, 2022, 10, 22228 Search PubMed.
- Z. Sun, Y. Su, A. Zhi, Z. Gao, X. Han, K. Wu, L. Bao, Y. Huang, Y. Shi, X. Bai, P. Cheng, L. Chen, K. Wu, X. Tian, C. Wu and B. Feng, Nat. Commun., 2024, 15, 4252 Search PubMed.
- J. Peng, Y. Su, H. Lv, J. Wu, Y. Liu, M. Wang, J. Zhao, Y. Guo, X. Wu, C. Wu and Y. Xie, Adv. Mater., 2023, 35, 2209365 CrossRef CAS PubMed.
- P. Wang, Y. Zhao, R. Na, W. Dong, J. Duan, Y. Cheng, B. Xu, D. Kong, J. Liu, S. Du, C. Zhao, Y. Yang, L. Lv, Q. Hu, H. Ai, Y. Xiong, V. S. Stolyarov, S. Zheng, Y. Zhou, F. Deng and J. Zhou, Adv. Mater., 2024, 36, 2400655 Search PubMed.
- K. Yang, Y. Zhou, Y. Ma and H. Wu, J. Mater. Chem. C, 2025, 13, 4549–4556 Search PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse, J. Hafner and G. Kern, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 8551 CrossRef.
- K. Burke, M. Ernzerhof and J. P. Perdew, Phys. Rev. Lett., 1997, 78, 1396 Search PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- S. Li, X. Bo, L. Fu, C. Liu, F. Li and Y. Pu, Appl. Phys. Lett., 2024, 124, 172407 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635 Search PubMed.
- X. Xu, Y. Ma, T. Zhang, C. Lei, B. Huang and Y. Dai, Nanoscale Horiz., 2020, 5, 1386–1393 Search PubMed.
- H. Lou and G. Yang, J. Mater. Chem. A, 2023, 11, 6394 RSC.
- B. Zhang, X. Fu, L. Song and X. Wu, Carbon, 2021, 172, 122 CrossRef CAS.
- W. Zhang, H. Lou and G. Yang, J. Phys. Chem. Lett., 2023, 14, 11036 CrossRef CAS PubMed.
- F. Mouhat and F.-X. Coudert, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- T. Nian, Z. Wang and B. Dong, Appl. Phys. Lett., 2021, 118, 033103 CrossRef CAS.
- S. Bertolazzi, J. Brivio and A. Kis, ACS Nano, 2011, 5, 9703 CrossRef CAS PubMed.
- S.-B. Chen, S.-D. Guo, W.-J. Yan, X.-R. Chen and H.-Y. Geng, Phys. Chem. Chem. Phys., 2024, 26, 3159 RSC.
- T. P. T. Linh, N. N. Hieu, H. V. Phuc, C. Q. Nguyen, P. T. Vinh, N. Q. Thai and N. V. Hieu, RSC Adv., 2021, 11, 39672 Search PubMed.
- X. Yang, L. Lin, X. Guo and S. Zhang, Small, 2024, 20, 2404000 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- J. Li, Nano-Micro Lett., 2022, 14, 112 CrossRef CAS PubMed.
- Y. Pi, Z. Qiu, Y. Sun, H. Ishii, Y.-F. Liao, X. Zhang, H.-Y. Chen and H. Pang, Adv. Sci., 2023, 10, 2206096 CrossRef CAS PubMed.
- Z. Wang, J. Li, S. Yuan, J. Yang, Z. Jin, X. Tan, J. Dang, W. Mu, G. Li and Q. Wang, Chem. Eng. J., 2024, 497, 154650 Search PubMed.
- J.-C. Liu, F. Luo and J. Li, J. Am. Chem. Soc., 2023, 145, 25264–25273 CrossRef CAS PubMed.
- X. Yao, J. Guo, Z. Wang, G. Qian, X. Wang, D. Wang and X. Gong, JACS Au, 2025, 5, 3926 CrossRef CAS PubMed.
- P. B. Calio, C. Li and G. A. Voth, J. Am. Chem. Soc., 2021, 143, 18672–18683 CrossRef CAS PubMed.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- J. Rossmeisl, E. Skúlason, M. E. Björketun, V. Tripkovic and J. K. Nørskov, Chem. Phys. Lett., 2008, 466, 68–71 CrossRef CAS.
- Q. Tang and D.-e. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- X. Liu, Y. Guo, F. Ning, Y. Liu, S. Shi, Q. Li, J. Zhang, S. Lu and J. Yi, Nano-Micro Lett., 2024, 16, 111 CrossRef CAS PubMed.
- Y. Luo, Y. Guan, G. Liu, Y. Wang, J. Li and L. Ricardez-Sandoval, ACS Catal., 2024, 14, 2696–2708 CrossRef CAS.
- X. Du, J. Huang, J. Zhang, Y. Yan, C. Wu, Y. Hu, C. Yan, T. Lei, W. Chen, C. Fan and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 4484 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71 CAS.
- Z. Feng, W. T. Hong, D. D. Fong, Y.-L. Lee, Y. Yacoby, D. Morgan and Y. Shao-Horn, Acc. Chem. Res., 2016, 49, 966 CrossRef CAS PubMed.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruña, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317 CrossRef CAS.
- Z. Fang, S. Li, Y. Zhang, Y. Wang, K. Meng, C. Huang and S. Sun, J. Phys. Chem. Lett., 2024, 15, 281 CrossRef CAS PubMed.
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4, 3966 RSC.
- Y.-L. Lee, D. Lee, X. R. Wang, H. N. Lee, D. Morgan and Y. Shao-Horn, J. Phys. Chem. Lett., 2016, 7, 244 CrossRef CAS PubMed.
- N. Mu, T. Bo, Y. Hu, R. Xu, Y. Liu and W. Zhou, Chin. J. Catal., 2024, 63, 244–257 CrossRef CAS.
- K. An, Z. Yu, H. Bai, D. Liu, L. Qiao, X. Lv, L. Shao, J. Feng, Y. Cao, L. Li, Z. Wen, S. Chen, Z. Pan, S. Wang and H. Pan, J. Mater. Chem. A, 2024, 12, 9672–9680 RSC.
- H. Qiu, T. Yang, J. Zhou, K. Yang, Y. Ying, K. Ding, M. Yang and H. Huang, J. Mater. Chem. A, 2023, 11, 7034–7042 RSC.
- J. Wang, J. Lu, X. Zhao, G. Hu, X. Yuan, J. Ren and S. Qi, J. Catal., 2026, 454, 116664 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |