DOI:
10.1039/D5SC07763J
(Edge Article)
Chem. Sci., 2026,
17, 8028-8042
Heavy-metal free near infrared photoredox catalysts in cancer phototherapy
Received
7th October 2025
, Accepted 23rd February 2026
First published on 25th February 2026
Abstract
Herein, five new far red/near infra-red (NIR) heavy-metal free photosensitizers (PSs) were developed by donor modulation of the planar perylenimide (PI) core, exhibiting large Stokes shifts of 213–270 nm. The dimethyl-enabled aggregation-induced enhanced emission (AIEE) and tunable AIEE/AIE behaviors of the RPI PSs aided precise and efficient superoxide (O2˙−) generation for cancer phototherapy, viz.R = Ph (PhPI), PhNH2 (ANPI), PhN(CH3)2 (DMPI), –PhN(Ph)2 (TPPI), PhN(BiPh)2 (BPPI) to obtain distinct NIR emitters and NIR photoredox catalytic properties. Notably, the solid state NIR emissive DMPI, TPPI and BPPI showed far-red/NIR AIEE, AIE and AIEE behaviors in aqueous media, whereas PhPI and ANPI displayed aggregation-caused quenching (ACQ) effects. A key discovery is the dimethyl-induced transformation of ACQ-to-AIEE in DMPI, enabling a very rare and unusual feature of inducing NIR AIEE properties in the PS. Moreover, the ACQ molecules PhPI and ANPI generate O2˙−/˙OH (type-I PS/photoredox) efficiently, DMPI exhibits NIR AIEE photoredox characteristics for O2˙− (type-I PS) generation, TPPI illustrates far-red AIE photoredox/type-II PS for O2˙−/1O2 (singlet oxygen quantum yield, ΦΔ = 0.59 in aqueous media), while BPPI demonstrated far-red AIEE photoredox for O2˙− in cancer cells. These systems highlight the diverse optical and therapeutic properties obtained by carefully varying the donor moiety in the RPIs. The most prominent dimethyl-induced NIR AIEE design strategy in DMPI reveals an exceptional heavy metal-free (NIR AIEE/NIR) photoredox catalyst PS, offering precise and efficient electron transfer for O2˙− production. Importantly, the NIR photoredox catalysts are rarely reported, and the introduction of NIR AIEE photoredox sensitizers expands the scope of current photoredox research. The photocatalytic superoxide generator TPPI, developed via triplet-ground-state splitting energy modulation, induces significant cancer cell death through a partial O2-recycling pathway involving Haber–Weiss/Fenton reactions.
Introduction
Phototherapy has emerged as a noninvasive alternative, leveraging light-activated PSs to generate ROS, particularly singlet oxygen (1O2) or type-I radicals (O2˙−/˙OH).1–3 Type-I phototherapy, favorable for hypoxic tumors, involves electron transfer from the triplet PS to molecular oxygen (3O2), generating O2˙− through superoxide disproportionation.4–6 In this photocatalytic strategy, O2˙− is generated upon photon excitation, followed by superoxide dismutase (SOD)-mediated disproportionation and Haber–Weiss/Fenton reactions to produce ˙OH. Through this process, a partial O2-recycling mechanism is established, enabling sustained oxidative stress and effective cancer cell death.5,7–9 Herein, the planar PI core was selected for its high fluorescence, chemical stability, biocompatibility, and thermal resilience, making it suitable for biomedical applications.10,11 Despite advances in NIR PS design, particularly donor–acceptor tuning and intersystem crossing (ISC) enhancement, major hurdles still remain due to the detrimental charge transfer characteristics at higher concentrations and the dark toxicity associated with heavy atoms for enhancing spin–orbit coupling (SOC).8,12,13
This study introduces a series of five newly synthesized peri-functionalized PI based PSs with unique photophysical properties, biocompatibility, and remarkable chemical and thermal stability, making them suitable for therapeutic and clinical applications.10,11 These five PSs include RPI [2-hexyl-8-phenyl-1H-benzo[5,10]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (PhPI)/8-(4-aminophenyl)-2-hexyl-1H-benzo[10,5]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (ANPI)/8-(4-(dimethylamino)phenyl)-2-hexyl-1H-benzo[10,5]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (DMPI)/8-(4-(diphenylamino)phenyl)-2-hexyl-1H-benzo[10,5]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (TPPI)/8-(4-(di([1,1′-biphenyl]-4-yl)amino)phenyl)-2-hexyl-1H-benzo[10,5]anthra[2,1,9-def]isoquinoline-1,3(2H)-dione (BPPI)], each possessing donor functionality with minor variations, attached to the PI core (Fig. 1). These RPI PSs were carefully designed and synthesized to modulate diverse properties, including far-red/NIR AIE/AIEE, higher Stokes shift in the range of 213–270 nm, distinct PS (type-I/dual type/type-II), and precise (NIR AIEE/NIR) photoredox catalysts that produce various ROS (˙OH/O2˙−). This work conceptualizes unique dimethyl-induced NIR AIEE characteristics in the planar PI core that arise from steric crowding around the pendant phenyl unit, which transforms ACQ into AIEE features in DMPI. Thus, DMPI demonstrated NIR AIEE photoredox catalytic properties (O2˙−, type-I PS) while TPPI exhibited type-II PS behavior and ΦΔ of 0.59 in aqueous media. TPPI and BPPI PSs exhibited solid state NIR emission, while TPPI showed dual type PS/photoredox (1O2 and O2˙−) behavior and BPPI was a type-I (O2˙−) PS/photoredox catalyst for effective cancer phototherapy. This work also hypothesized the ground-triplet state splitting energy for meticulous NIR photoredox catalyst formulation. Thus, this approach offers for the first time a promising platform for the development of heavy-metal-free design principles for advancing distinct (NIR/NIR AIEE) photoredox systems that generate precise ROS (O2˙−). Additionally, this work also revealed a unique dimethyl-induced effect in NIR AIEE design that enabled diverse structure based optical tuning (Fig. 1 and Tables S1, S2). Notably, while NIR photoredox catalysts are rare, the development of NIR AIEE photoredox catalysts represents a significant conceptual advancement. TPPI is designed via an intramolecular triplet-ground-state splitting energy modulation strategy; it functions through a partial O2-recycling mechanism in which O2˙− drives Haber–Weiss/Fenton reactions, enabling effective cancer cell death.
 |
| | Fig. 1 The graphical representation of various RPIs illustrates far-red/NIR condensed state emitters, large Stokes shift (213–270 nm) tuning, and NIR/NIR AIEE photoredox catalytic activity for O2˙− generation and diverse PS functionalizations (type-I/dual type/type-II) for cancer phototherapy (IC50 = 20.5 µM). The graphic represents dimethyl-induced NIR AIEE behavior, and highlights a triplet-ground-state splitting modulation strategy that enables a partial O2-recycling mechanism, where O2˙− drives Haber–Weiss/Fenton reactions that could be applicable for effective cancer cell death even under hypoxia (≤2% O2). | |
Results and discussion
Design, synthesis, and characterization of RPIs
This study explores the condensed state optical and NIR photoredox catalytic properties of novel RPIs with minor donor unit variation at the peri-position of the acceptor PI core, viz, RPI: PhPI/ANPI/DMPI/TPPI/BPPI, that have been carefully synthesized from PDA via hexylamine condensation, bromination, and Suzuki coupling with good yields. Detailed synthetic procedures and characterization data are provided in Schemes 1 and S1–S3 (SI).
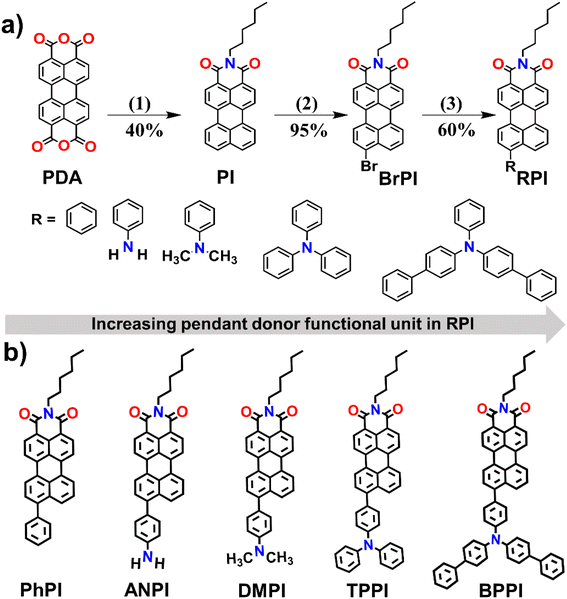 |
| | Scheme 1 (a) Synthetic method employed for preparing RPIs (PhPI/ANPI/DMPI/TPPI/BPPI) with increasing donor unit at the peri-position of the PI core. (b) Chemical structures of the synthesized RPIs [(1) hexyl amine, H2O, 20 h, (2) chlorobenzene, Br2, 4.5 h, (3) Pd (0), THF/H2O, values below the arrow are the yields of corresponding products]. | |
Photophysical behaviors
The optical properties of the newly designed RPIs were investigated using UV-vis absorption and photoluminescence (PL) spectroscopy in solution (DMSO), aggregated (DMSO/H2O) and solid states (Fig. 2 and Tables 1, S3). The absorption spectra revealed that increasing the donor functional unit in the peri-position significantly influenced π-conjugation within the PI core (Fig. 2a and Table 1). Among them, ANPI (absorption maxima, λabs,max = 550 nm)/DMPI (λabs,max = 550 nm) exhibited a 30/50 nm red-shifted λabs,max due to their enhanced delocalization, compared to PhPI (λabs,max = 500 nm). Further increasing the donor unit at RPI, viz.TPPI (λabs,max = 527 nm)/BPPI (λabs,max = 525 nm) exhibited a 23/25 nm blue shifted λabs,max relative to DMPI. This peculiar shift is attributed to reduced electronic communication, likely due to their steric hindrance and spatial separation of the HOMO/LUMO, as supported by further DFT analyses. Corresponding emission maxima (λem,max) showed trends consistent with λabs,max. ANPI (λem,max = 603) exhibited a 42 nm red-shifted λem,max, compared to PhPI (Fig. 2b, b′ and Tables 1, S3). DMPI (λem,max = 588 nm) showed less pronounced λem,max, while TPPI/BPPI demonstrated 42/39 nm blue shifted λem,max compared to PhPI, despite having stronger donor units. This inconsistency suggests reduced electronic communication in the solution state. The solution state λem,max, spanning yellow-green to red, has been presented in the CIE chromaticity diagram (Fig. 2b′ and Table S3). However, RPIs demonstrated prominent red-shifted λem,max of ∼100/150 nm in their aggregated/solid state (Fig. 2c, d, and Table 1). DMPI displayed NIR λem,max at 720/770 nm in its aggregated/solid state, which arises from its effective molecular packing. TPPI/BPPI exhibited far-red/NIR λem,max at 686/713 nm and 675/716 nm, respectively, in their aggregated/solid state. The CIE plot exhibited far-red/NIR colors in the aggregated-state (Fig. 2c′, d′ and Tables 1, S3). DMPI/TPPI/BPPI showed significant 50/27/41 nm red-shifted λem,max in their solid state compared to the aggregated state due to their distinct molecular arrangements (Fig. 2e and Table 1). In contrast, PhPI/ANPI showed minimal shifts in their condensed state, indicating stable conjugation. This distinct molecular arrangement was further analyzed using excitation-emission matrix (EEM) spectroscopy. Under white light and 365 nm UV illumination, visible appearance differences in powder form were observed. PhPI/ANPI appeared colorless due to their non emissive nature, while DMPI also lacked visible color owing to its NIR emission, that requires NIR excitation. TPPI/BPPI showed a distinct far-red color optical appearance (Fig. 2f and g).
 |
| | Fig. 2 Photophysical properties of the RPIs (PhPI/ANPI/DMPI/TPPI/BPPI): (a) normalized UV-vis absorbance spectra in DMSO (solution state, 100 µM). (b) PL spectra of RPIs in solution state. (b′) CIE chromaticity diagram for solution state fluorescence color. (c) Normalized PL spectra at 99% fw in DMSO. (c′) CIE coordinates in the aggregated state. (d) Solid state normalized PL spectra. (d′) CIE chromaticity diagram for solid state emission color. (e) Comparison of PL spectra in aggregated and solid states. (f and g) Digital photographs of the solid RPIs in powder form under daylight and UV excitation (λex = 500 nm). [a and s: aggregated and solid state, respectively; λex = 500 nm. Inset in CIE: 1–5 = PhPI/ANPI/DMPI/TPPI/BPPI]. | |
Table 1 Summary of the photophysical and PS/photoredox catalytic information of the RPIs
| Photosensitizers |
λ
abs (nm) |
λ
ex (nm) |
λ
em (nm) |
λ
em (nm) |
λ
em (nm) |
Stokes-shift (nm) |
Φ
PL
|
E
g (eV) |
Φ
Δ
|
ΔES1–T1 (eV) |
SOC (cm−1) |
ΔES0–T1 (eV) |
|
A–E |
S–E |
|
λ
ab,max, λex and λem,max represent absorption, excitation, and emission wavelength maxima, respectively, in the solution state.
Aggregated and solid state λem,max.
Stokes-shift calculated between the aggregated-state λem,max and λex (A–E), whereas (S–E) represents the Stokes-shift between the solid state λem,max and λex.
Φ
PL indicates the PL quantum yield in the aggregated state.
E
g demonstrates gaseous-state band-energies.
Φ
Δ represents the singlet-oxygen quantum yield.
ΔEST is the first singlet and triplet state energy gap.
SOC between the singlet and triplet states. [p = ΦPL at 60% fw]. ΔES0–T1 is the intramolecular triplet-ground state splitting energy. g = not observed.
|
|
PhPI
|
500 |
500 |
580 |
646 |
646 |
146 |
146 |
0.0387 |
2.66 |
0.0054 |
1.182 |
0 |
1.165 |
|
ANPI
|
530 |
500 |
603 |
620 |
620 |
117 |
117 |
0.0693 |
2.566 |
0.0045 |
1.083 |
0 |
1.079 |
|
DMPI
|
550 |
500 |
588 |
720 |
770 |
220 |
270 |
0.196, 0.044p |
2.480 |
g |
0.623 |
0.447 |
1.002 |
|
TPPI
|
527 |
500 |
561 |
686 |
713 |
186 |
213 |
0.0534, 0.098p |
2.410 |
0.592 |
0.639 |
0.141 |
2.532 |
|
BPPI
|
525 |
500 |
564 |
675 |
716 |
175 |
216 |
0.033, 0.0999p |
2.340 |
0.025 |
0.522 |
0.141 |
0.871 |
Thus, by varying the donor units, the λem,max trend remained unaffected by the electronic properties of these functional units in their aggregated/solid state. This highlights that AIE properties are governed by functional units, suggesting that condensed state photophysical properties are independent of electronic effects.14–16 The functional units play pivotal roles, exerting varying steric constraints on the parent PI core, thereby manipulating its molecular organization and resulting in distinct condensed state emitters. Moreover, the RPIs exhibited exceptional Stokes shifts (117–270 nm), surpassing the widely used isoquinoline-based analogs like IQ-TPA (173 nm) and TPE-IQ-TPA (212 nm).17,18
ACQ to far-red/NIR AIE/AIEE transformation
To investigate the aggregation properties of the RPIs, UV-vis, and PL spectra were measured in 99% water fractions (fw) (Fig. 3, S1–S13 and Tables 1, S1–S10). At 0% fw, ANPI/DMPI/TPPI/BPPI displayed λabs,max at 530/550/527/525 nm, respectively, corresponding to π–π* transitions (Fig. S2, S3, and Table S4). PhPI showed two characteristic peaks at 483/500 nm, indicating π–π* and intramolecular charge transfer (ICT) from phenyl donor to the acceptor PI core. Furthermore, solvent polarity-dependent PL studies confirmed ICT nature in the RPIs (Fig. S5 and Tables S7, S8). DMPI exhibited a more pronounced ICT effect, comprising 38/56 nm red-shifted λabs,max/λem,max, respectively, while TPPI/BPPI showed 12/49 and 10/38 nm red-shifted λabs,max/λem,max in polar solvents, respectively. At 99% fw, PhPI/ANPI exhibited prominent ∼70/73 nm blue shifted λabs,max due to H-aggregation energized by pronounced π–π stacking of the acceptor PI core (Tables S4 and S5). Conversely, DMPI exhibited 10 nm red-shifted λabs,max (J-type aggregation). Importantly, TPPI/BPPI showed distinct spectral shifts at 99% fw. This peculiar spectral behavior observed in the concentration-dependent studies in DMSO (100 µM–1 mM), revealed stable λabs,max for PhPI/ANPI, whereas DMPI/TPPI/BPPI exhibited 12/1/3 nm red-shifted λabs,max (J-type aggregation) (Fig. S4 and Table S6). At 0% fw, PhPI/ANPI/DMPI/TPPI/BPPI showed corresponding λem,max at 580/603/588/561/564 nm, respectively (Fig. 3 and S1). At 99% fw, PhPI/ANPI showed ACQ behavior consisting of quenched and red-shifted λem,max at 646/620 nm (Fig. 3a–a′′, S1 and Table 1). Although DMPI/TPPI/BPPI showed less pronounced or blue shifted λem,max compared to PhPI/ANPI, DMPI exhibited NIR AIEE features (λem,max = 720 nm) while TPPI/BPPI displayed far-red AIE/AIEE characteristics (λem,max = 686/675 nm) (Fig. 3b–b′′, c–c′′, d–d′′ and Table 1). The distinct H- and J-aggregation behaviors observed for these RPI derivatives can be rationalized based on Kasha's exciton coupling model for one-dimensional molecular assemblies, in which positive and negative excitonic coupling lead to blue- and red-shifted optical spectra relative to the monomer, corresponding to H- and J-aggregates. Small organic chromophores typically form H- or J-type aggregates depending on the relative orientation of their transition dipole moments, and the aggregation mode strongly governs the position and shape of their absorption bands. Compared with isolated monomers in the diluted solution, a hypsochromic (blue) shift is characteristic of H-aggregation arising from cofacial π–π stacking, whereas a bathochromic (red) shift indicates J-aggregation associated with slipped or head-to-tail molecular arrangements.19,20 In H-aggregates, positive coulombic (exciton) coupling produces a manifold of excited states in which the highest energy state carries most of the oscillator strength. Because fluorescence typically occurs from the lowest excited state (Kasha's rule), radiative decay is suppressed in H-aggregates, favoring nonradiative pathways such as internal conversion and intersystem crossing.21 In contrast, J-aggregates are characterized by negative excitonic coupling that concentrates oscillator strength in the lowest-energy exciton state, thereby preserving or even enhancing fluorescence. Consequently, controlling the aggregation mode provides an effective strategy to tune the photophysical and electronic properties of organic semiconductors.19,20,22,23 In this context, PhPI exhibits H-type aggregation, as evidenced by its pronounced blue shifted λabs,max (Fig. S3 and Tables S4, S5). In contrast, although BPPI displays invariant λabs,max at a lower concentration, it shows J-type aggregation behavior, as supported by concentration-dependent UV-vis absorption studies in both mixed solvents and DMSO (Fig. S3, S4 and Tables S4, S6). Specifically, in DMSO (100 µM–1 mM), PhPI shows an almost invariant λabs,max, whereas BPPI exhibits a gradual red shift of approximately 3 nm, consistent with J-aggregate formation (Fig. S4, and Table S6). These distinct spectral features highlight the fundamentally different aggregation modes of PhPI and BPPI, which has been further confirmed by the characteristic peak appearance for BPPI upon aggregation (Fig. 3a and d). Although TPPI exhibits a 27 nm blue shifted λabs,max, its J-aggregation is indicated by a 1 nm red-shifted λabs,max UV peak at higher concentrations, which subsequently leads to its promising characteristic peak appearance at 686 nm upon aggregation (Fig. 3c). In the present system, strong π–π interactions of the electron acceptor PI core promote H-type aggregation (Fig. S3 and Tables S4, S5). However, DMPI exhibits a distinct approximately 10/12 nm red-shifted λabs,max both at lower and higher concentrations, indicating a more prominent J-type aggregation character (Fig. S3, S4 and Tables S4, S6). This J-type packing mitigates the fluorescence quenching typically induced by strong π–π stacking of the PI core and, together with restriction of intramolecular rotation (RIR), leads to enhanced emission in the aggregated-state. The extent of J-aggregation among DMPI, TPPI, and BPPI follows the order DMPI > BPPI > TPPI, as reflected by their respective bathochromic λabs,max shifts of approximately 12, 3, and 1 nm in the aggregated state (considering the UV-vis spectra at higher concentrations as aggregation is the concentration dependent phenomenon, and likely affects the UV-vis spectra by involving aggregated molecules) (Fig. S4 and Table S6).15 This trend can be attributed to the steric and conformational effects of the donor units attached to the PI core. In DMPI, the flexible donor moiety enables greater segmental motion and closer intermolecular association, leading to aggregation-induced red-shifted emission and pronounced NIR AIEE behavior. In contrast, the bulkier donor units in TPPI and BPPI restrict segmental motion and hinder effective aggregation,
thereby suppressing large spectral red shifts and resulting in far-red AIE/AIEE emission. Previous studies have shown that solvent polarity and aggregation strongly influence the emission behavior of donor–acceptor luminogens.24–30 Interestingly, For BPPI, the λem,max is observed at 564 nm in pure DMSO (0% fw). As solvent polarity increases, the twisted ICT (TICT) state stabilizes, resulting in a red shift and a decrease in emission intensity up to 40% fw. Beyond this critical point, molecular aggregation restricts intramolecular motion and suppresses TICT formation, consequently, emissive J-type aggregates dominate the photophysical behavior with a new characteristic peak at 675 nm (far-red AIEE) (Fig. 3d–d′′).14 The decline of λem,max after aggregation was due to its agglomeration.31 Furthermore, the AIE/AIEE behavior of all the molecules was estimated by correlating PL photographs, and PL intensity versus fw profiles.14 Herein, PhPI and ANPI exhibited typical ACQ properties, as evidenced by decreased emission upon aggregation. In contrast, DMPI, TPPI, and BPPI displayed AIEE/AIE characteristics. When molecularly dispersed in DMSO, the luminophores are weakly or moderately emissive, and as the fw increases, emission initially decreases due to enhanced TICT formation. Beyond a critical fw, aggregation occurs and the RIR suppresses the nonradiative decay pathway, promoting radiative relaxation and resulting in enhanced emission. In solution, fluorescence is mainly governed by the rotational motion of the functional groups appended to the core, energized by their diverse electronic effects, whereas in the aggregated or solid state, intermolecular steric effects hinder these motions and favor radiative decay pathways (Fig. 3).15,32 Interestingly, BPPI exhibits weak red emission in the solid state, despite showing strong emission in DMSO (Fig. 2g and 3d′′). As shown in Fig. 2f and g, BPPI displays red emission in the solid state under 365 nm UV irradiation, which is distinctly different from its appearance under daylight. However, the solid-state emission is relatively weak, which can be attributed to its inefficient intermolecular packing interactions arising from the steric constraints imposed by the attached bulkier functional units on the parent PI core. In contrast, the strong emission observed in DMSO originates from the promising electronic effect of the appended group when the BPPI molecules are in an isolated state. The solid-state emission governed by the restricted packing and solution state emission dominated by intrinsic electronic transitions are consistent with previously reported luminogens.15,16,32 More interestingly, PhPI exhibits red emission in the solid state and yellow emission in DMSO; however, it becomes non-emissive upon aggregation (Fig. 2g and 3a–a′′). The solution state emission of PhPI arises due to its unique electronic constraints of the appended unit, where molecules are in a totally dispersed state. In the solid state, restricted intramolecular motion partially suppresses nonradiative decay pathways, leading to red emission. In contrast, in the aggregated-state, strong π–π stacking interactions between planar PhPI molecules facilitate exciton coupling and promote nonradiative relaxation pathways that could be due to its excited state planarity and increase reorganization energy, which further enhances internal conversion and suppresses radiative emission, thereby quenching fluorescence (ACQ effect).33–35 This effect clearly indicates a significant difference in reorganization energy and excited state planarity relaxation in solvent from solid in PhPI.
 |
| | Fig. 3 PL spectra of (a) PhPI, (b) DMPI, (c) TPPI, and (d) BPPI, at different fw in DMSO (100 µM, λex = 500 nm). (a′–d′) Plots of λem,max at various fw along with insets: (a′′–d′′) digital photographs under 365 nm UV irradiation of the corresponding RPI at 0/40/20/99% fw in DMSO with their corresponding chemical structures [daylight photographs of all the RPIs have been placed in Fig. S11 in the SI]. | |
Furthermore, the RPIs showed low ΦPL (0.03–0.2) at 99% fw due to ACQ or NIR characteristics that are responsible for the non-radiative decay process (Fig. S12, S13, Table 1 and eqn (S1)). DMPI/TPPI/BPPI demonstrated 22/11/10 nm blue shifted λab,max in fHex (99% hexane in CHCl3) (Fig. S6, S7 and Table S9). However, concentration-dependent λab,max of DMPI/TPPI/BPPI exhibited 3/2/1 nm red-shifted λab,max (J-type aggregation) (Fig. S8 and Table S10). At 0% fHex, DMPI/TPPI/BPPI revealed corresponding λem,max at 676/657/657 nm, followed by blue shifted λem,max enhancement at 570 nm in 99% fHex, though λab,max/λem,max shifts were less pronounced for TPPI/BPPI at 0% fHex due to reduced π-delocalization (showed similar characteristics with fw) (Fig. S9). Moreover, DMPI/TPPI/BPPI exhibited 123/130/134 nm hydrodynamic radii that are suitable for biomedical applications (Fig. S10).
EEM spectra
Excitation Emission Matrices (EEMs) are widely applied in systems requiring multi-component analysis, as they capture unique spectral signatures that are often referred to as molecular fingerprints for complex mixtures. In the present study, 3D EEM spectral scans were recorded for the representative RPIs over a concentration range of 100 µM–1 mM, revealing distinct fluorophore behaviors associated with each system (Fig. 4).36 At lower concentrations (100–500 µM), PhPI/ANPI predominantly exhibited a single emissive species, consistent with the presence of isolated fluorophores, whereas DMPI/TPPI/BPPI displayed dual fluorophore contributions. Thus, time-resolved photoluminescence (TRPL) measurements were performed, and the decay profiles were well fitted using a single-exponential function, which describes the relaxation of an isolated population of excited fluorophores according to eqn (1):| | | f(t) ≡ I(t)/I(0) = exp(−t/τD), | (1) |
where I(0) is the initial emission intensity determined by the concentration of excited donors, and τD represents the intrinsic excited state lifetime of the fluorophore (i.e., the inverse of the spontaneous emission rate constant). The observation of single-exponential fluorescence decay indicates the presence of a single dominant emissive species and confirms that emission originates from a well-defined excited state without significant contribution from multiple emissive pathways or heterogeneous emissive environments. Notably, for PhPI and ANPI, the fluorescence lifetimes show only negligible variation with concentration, suggesting that excited state relaxation converges into a single emissive state and that radiative decay proceeds through a unique fluorophoric pathway. Such behavior is consistent with systems in which aggregation does not introduce additional emissive traps or excimer-like species (Fig. S14).37–39 Notably, TPPI's intrinsic fluorophore was essentially non-emissive at these low concentrations, indicating its weak radiative decay under dilute conditions. Upon increasing the concentration, clear trends in spectral broadening and wavelength shifts emerged. PhPI/DMPI exhibited progressively broadened spectra, suggesting concentration-driven intermolecular interactions and aggregation. By contrast, ANPI retained a more symmetric profile with a pronounced blue shift in λex, indicative of reduced conjugation. At 1 mM, the behavior diverged more prominently: DMPI/TPPI/BPPI all developed asymmetric emission features with λex undergoing a distinct red-shift. In particular, TPPI displayed a dramatic enhancement in fluorophore intensity, surpassing that of the other RPIs, consistent with the activation of its otherwise quenched emissive unit under high concentration conditions. The contour plots further clarified these effects: red-shifted λex correlated with extended π-conjugation and stronger intermolecular electronic coupling, while blue shifted λex signal decreased conjugation or enhanced steric disruption of delocalization.36,40 Collectively, these observations indicate that the photophysical responses of the RPIs are strongly modulated by concentration-dependent aggregation, conjugation, and steric constraints imposed by pendant functional groups. Such effects govern the balance between emissive and non-emissive states, ultimately dictating the spectral fingerprints captured in EEM analyses.
 |
| | Fig. 4 (a1–e4) 2D EEM contour plots of the RPIs from lower to higher concentrations (left: 100 µM; middle: 300 µM and 500 µM; right: 1 mM) in DMSO [inset: arrow signifies the PL intensity]. | |
Electronic properties
DFT analysis (B3LYP/6-31G (d, p)) revealed decreasing HOMO–LUMO gaps (Eg) with stronger donor units in the RPIs (Fig. 5, S15–S18 and Table 1): PhPI (2.660 eV) > ANPI (2.566) > DMPI (2.480) > TPPI (2.410) > BPPI (2.340 eV) (Fig. 5 and Table 1). LUMOs were confined to the PI core, while HOMOs extended over donor units, indicating strong push–pull interactions and enhanced π-conjugation, especially in DMPI/TPPI/BPPI. This correlated with red-shifted λabs,max and far-red/NIR λem,max. However, TPPI/BPPI showed slightly reduced π-delocalization, leading to less pronounced or blue shifted λabs,max/λem,max/Stokes shifts at their dispersed state. Eg decreased more in polar solvents (DMSO/water) than in nonpolar ones (CHCl3/hexane), which also showed blue shifted λem,max in hexane due to reduced π-conjugation (Fig. S15–S18).41,42
 |
| | Fig. 5 Optimized ground state band energies (S0) in eV of the RPIs in the gaseous state, calculated using DFT/B3LYP with the 6-31G (d, p) basis set (Gaussian 16). | |
Theoretical calculation
Increasing the donor unit leads to reduced Eg in DMPI/TPPI/BPPI (2.480/2.410/2.340 eV), which aligns well with their optical behavior (Fig. S19 and Tables 1, S11–S13).12,43,44 These derivatives exhibited small ΔES1–T1 of 0.623/0.639/0.522 eV, comprising significant SOC (ζ(S1, T1): 0.447/0.141/0.141 cm−1) that enabled efficient S1–T1 transitions. In contrast, although PhPI/ANPI showed 0 cm−1ζ (S1,T1), which leads to favor the S1–T1 transitions, yet they displayed prominent ζ (S2,T2) of 3.714/0.435 cm−1, containing a lower ΔES2–T2 of 0.347/0.312 eV. DMPI/TPPI/BPPI also showed notable ζ (S2,T2) of 0.700/0.282/0.387 cm−1 with ΔES2–T2 of 0.637/0.628/0.703 eV, respectively. Thus, DMPI/TPPI/BPPI confirmed favorable ISC characteristics for PS properties.
Singlet oxygen (type-II ROS) detection
Due to the substantial SOC and high T1 population in the RPIs, ΦΔ was evaluated using 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) in 99% fPBS (99% PBS in DMSO) under white light irradiation (Fig. 6a, S20, S21, S22a, Tables 1, S14 and eqn (S2)). TPPI showed the highest ΦΔ of 0.592 (type-II), while BPPI had a moderate ΦΔ of 0.025. Other RPIsviz.PhPI/ANPI/DMPI demonstrated no detectable 1O2 generation. Notably, TPPI exhibited a high ΦΔ in aqueous media, though higher values (0.91/1.0/1.1) were reported for MeTTPy/NI-PSs/LOCK in organic solvents (Table S14).
Total ROS generation evaluation
Total ROS generation by the RPIs was assessed using 2,7-dichlorodihydrofluorescein (DCFDA) under white light in fPBS (Fig. 6b and S22b–d, S23). TPPI/BPPI/DMPI showed 130/110/30-fold PL enhancement after 30 min of white light irradiation, while only 8-fold enhancement was observed for PhPI/ANPI. TPPI/BPPI/DMPI induced 2.5 × 105/2.0 × 105/1.8 × 105 PL enhancement within 5 minutes, confirming both type-I and type-II ROS. Thus, DMPI/TPPI/BPPI NIR/Far-red AIEE/AIE PSs show efficient ROS generation compared to recently reported PSs (Table S2). Efficient ROS production by TPPI/BPPI is attributed to strong donor units attached to these RPIs, which exert promising ICT characteristics, correspondingly lowering the ΔEST (e.g., BPPI: 0.522 eV) and enhancing T1 population via ISC (Fig. S19 and Table 1).45BPPI's low ΦPL (0.03) further supports ISC-driven ROS (O2˙−) generation.45
 |
| | Fig. 6 (a) Plots of ABDA (100 µM) decay rates at λabs,max of 380 nm by the different RPI PSs (100 µM). (b) PL plots of the DCFDA indicator in the presence of distinct RPIs. A0 and A are ABDA absorbances at λabs,max of 380 nm, whereas I0 and I are PL of the indicator at λem,max of 521 nm in the presence and absence of white light irradiation. (c) ESR signals in 99% fPBs. (d) PL spectra of TA in the presence of the RPIs at 528 nm. (e) Plots of zeta potential at different pH levels of the RPIs. (f) Ev and Ec of the RPIs at pH 5.6. The energy scale is represented in relation to NHE. (g) ΔG variations of PhPI/ANPI/DMPI/TPPI/BPPIvia ORCA 5.0 SOCME software at the B3LYP DEF2-SVP level. | |
Radical (type 1 ROS) detection
The free radical generation of RPI was studied via ESR using TEMP as a 1O2 trap in 99% fPBS under white light (Fig. 6c). Only TPPI showed a detectable ESR signal, confirming 1O2 generation. Terephthalic acid (TA) probing revealed significant HO˙ production only by ANPI (Fig. 6d).43 These findings suggest a dominant type-I mechanism via O2˙− generation through electron transfer from T1 to 3O2. Following the reported method,46 the points of zero zeta potential (PZZP) of the PIs were estimated from the zeta potential versus pH plots (Fig. 6e, f, S24, S25, Tables S15–S17 and eqn (S3–S6)). Initially, the aqueous solutions of the PIs were neutral (pH 7). Zeta potential measurements showed a transition from negative to positive values as the pH decreased from alkaline (pH 9) to acidic (pH 1). As shown in Fig. 6e, the PZZP values of PhPI, ANPI, DMPI, TPPI, and BPPI were 1.49, 0.103, 17.3, 1.126, and 1.23, respectively (Table S17). The valence band energy (Ev) was determined from Ultraviolet Photoelectron Spectroscopy (UPS) measurements. The conduction band energy (Ec) was estimated using the optical band gap obtained from the onset of UV-visible absorption spectra. The Ec relative to the normal hydrogen electrode (NHE) were calculated using eqn (S3) and (S4). The calculated Ev and Ec values for the RPIs are summarized in Table S17. Furthermore, the Ev and Ec at pH 5.6, corresponding to the PZZP, were obtained using eqn (S5) and (S6). The Ec for PhPI/ANPI/DMPI/TPPI/BPPI were −1.20249/−0.354323/−1.1397/−2.113966/−1.07783 eV, respectively, at pH 5.6 enabling electron transfer to 3O2.32 However, Ev for PhPI/DMPI/TPPI/BPPI were 1.12/0.02/0.09/1.10 eV, which are below the redox potentials for HO˙ (2.20 eV) and 1O2 (1.88 eV), making their generation unlikely – except for TPPI (1O2 generation explained via ground-triplet state energy splitting, ΔES0–T1) and ANPI (HO˙ generate due to its Ev closet to 2.20, Ev ≈ 1.90 eV). Gibbs free energy variations (ΔG) from DFT (ORCA 5.0) indicate favorable intermolecular electron transfer (photoredox catalyst), especially for ANPI (ΔGOH = – 413.93 kcal mol−1), confirming its potential HO˙ production (Fig. 6g and Tables S18–S31).43
Photoredox catalyst for O2˙− generation
Light-triggered O2˙− generation is vital for hypoxia-compatible phototherapy, as it enables 3O2 on empirical data and rarely produces O2˙− (Fig. 7).5,8,9,43–45,47 Efficient O2˙− generation requires rapid electron transfer from 3PS* to 3O2 and ΔES0–T1 smaller than 3O2/1O2 energy gap (∼1.610 eV) to prevent 1O2 formation (Fig. 7d).45 Although all the RPIs (PhPI/ANPI/DMPI/TPPI/BPPI) exhibit T1 energies (1.495/1.487/1.478/1.473/1.469 eV) above 0.98 eV, which is sufficient for 3O2 sensitization, they fail to produce 1O2.48 Thus, there is an urgent need for ΔES0–T1 calculation. PhPI/ANPI/DMPI/BPPI showed ΔES0–T1 of 1.165/1.079/1.002/0.871 eV, which are below the 1.610 eV threshold required for 1O2 generation that favors O2˙− formation via ISC (Tables 1 and S32). Since type-II energy transfer requires ∼94.5 kJ mol−1, this becomes thermodynamically unfavorable, consisting of a smaller ΔES0–T1.6,45,47TPPI remains an exception that generates 1O2 favorably due to higher ΔES0–T1 of 2.532 eV. These findings confirm that tuning of ΔES0–T1 in a D-A system enables selective O2˙− generation, supporting their role as efficient photoredox catalysts (Fig. S26).
 |
| | Fig. 7 (a) ESR plots of DMPO for type-I ROS O2˙− categorization in the presence of RPI PSs (PhPI, ANPI, DMPI, TPPI, or BPPI) after 20 min of white light irradiation in dry acetonitrile solution. (b) The variation of UV-vis spectra of XTT at 470 nm after being illuminated with white light for 30 min in the presence and absence of PhPI, ANPI, DMPI, TPPI, or BPPI in 99% fPBS. (c) DHR 123 for O2˙− detection in the presence of PhPI, ANPI, DMPI, TPPI, or BPPI after white light excitation at various times in 99% fPBS. (d) Schematic representation of an RPI to produce O2˙− under white light excitation. [RPI] = 100 µM. | |
ESR with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) showed six-line signals, confirming O2˙− adducts (Fig. 7a).45,47–49 2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assays revealed increased 470 nm absorption upon irradiation (Fig. 7b and S27), while the PL of dihydro rhodamine 123 (DHR123) at 527 nm confirmed O2˙− production (Fig. 7c and S28).5,8,46,49 After 30 min of light exposure, the PL increased for PhPI/ANPI/DMPI/TPPI/BPPI by 3/3/2/4/8-fold, validating efficient O2˙− generation by these RPI photoredox catalysts (Fig. 7d).
White light-induced phototherapy performances
DMPI/TPPI/BPPI demonstrated efficient ROS generation and photocatalytic redox activity for in vitro phototherapy on HeLa cells (Fig. 8a). MTT assays revealed potent photo-cytotoxicity for TPPI (IC50 = 20.5 µM) under 30 min white light illumination, with complete cell eradication at 100 µM despite slight dark toxicity. BPPI shows dark toxicity after 10 µM and completely eradicates the HeLa cell at 100 µM under light treatment. DMPI showed minimal light-induced cytotoxicity due to lower ROS output, requiring NIR activation. Furthermore, photoirradiation experiments were performed on HeLa cells using a 20 µM concentration of the materials over different time intervals. TPPI exhibited the highest cell killing efficiency, reaching approximately 65%, which can be attributed to its prominent PS properties under white light irradiation (Fig. S29). In contrast, BPPI and DMPI showed significantly lower cell killing efficiency, likely due to their reduced PS activity. Moreover, field-emission scanning electron microscopy (FESEM) was performed on the primary material TPPI and the comparative material ANPI to investigate their morphological evolution under light irradiation. The FESEM images reveal that TPPI undergoes pronounced morphology changes upon light irradiation, as evidenced by a significant reduction in particle size from approximately 200 nm to well-defined nanoparticles of about 100 nm or smaller, accompanied by a transformation from more uniform and ordered nanostructure to a irregular aggregates (Fig. S30–S33). In contrast, ANPI neither exhibits any notable morphological transformation nor does it show a significant change in nanoparticle sizes before and after the light irradiations. These observations provide strong visual evidence of the light responsive structural reorganization (Fig. S34 and S35). Fluorescence imaging further confirmed TPPI's phototherapeutic efficacy.
 |
| | Fig. 8 (a) Cytotoxic study of HeLa cells post treatment with various concentrations of DMPI/TPPI/BPPI along with 30 min of white light illumination. Fluorescence microscope images of HeLa cells after treatment with (b) DMPI/TPPI/BPPI PSs under normoxia, loaded with PI (4 µM, dead cell marker), calcein-AM (2 µM, live cell marker), and DCFDA (10 µM, ROS indicator) [concentration: DMPI = 20 µM, TPPI = 20 µM, and BPPI = 25 µM; scale bar: 100 µm]. | |
Live/dead cell dual staining and intracellular ROS generation
Calcein-AM/PI staining and DCFDA imaging confirmed effective ROS generation by TPPI in HeLa cells under white light (Fig. 8b), with green fluorescence indicating elevated ROS levels. The treated cells showed morphological changes, such as shrinkage, reduced size, and collapse, supporting TPPI-induced photodynamic damage.
Plausible therapeutic mechanism of action for O2˙− mediated phototherapy
Oxidative stress is defined as an imbalance between reactive oxygen/nitrogen species (ROS/RNS) and the cellular antioxidant defense system, which includes both enzymatic and nonenzymatic antioxidants. Sustained oxidative stress leads to biomolecular damage and contributes to various pathological conditions, including carcinogenesis and neurodegenerative disorders.50–53 In the present study, TPPI induced photodynamic treatment exhibited pronounced morphological changes in HeLa cells, such as cell shrinkage, reduced cell volume, and collapse of cellular structures, indicating severe oxidative damage (Fig. 8b). This could be due to the partial O2-recycling mode of action, allowing oxidative stress to be efficiently triggered even at low oxygen tension, thereby promoting photodynamic damage in cancer cells.5,45 These effects are primarily attributed to light-triggered O2˙− generation, which subsequently participates in Fenton and Haber–Weiss reactions to yield highly reactive ˙OH. This cascade enables a partial O2-recycling mechanism, allowing efficient oxidative stress induction even under low oxygen tension.50,51 Herein, the photoexcited photosensitizer (TPPI) transfers an electron or proton to triplet oxygen (3O2) to generate O2˙−. The resulting O2˙− undergoes SOD-mediated disproportionation to form H2O2 and regenerate 3O2 (reaction 2). Subsequently, H2O2 reacts with Fe2+via the Fenton reaction to produce ˙OH and Fe3+, while Fe3+ can be reduced back to Fe2+ by O2˙−, regenerating 3O2 (reaction 3). In parallel, the Haber–Weiss reaction provides an additional pathway for ˙OH production and 3O2 regeneration (reaction 4). To verify this mechanism, Fenton–Haber–Weiss reactions were performed following reported protocols using TPPI under 30 min light irradiation, and the reaction products were analyzed by liquid chromatography-mass spectrometry (LC-MS). The enhancement of LC-mass peak of catechol at 110 [M] and 2,3-dihydroxybenzoic acid/2,5-dihydroxybenzoic acid at 155 [M]+ under light irradiation, the characteristic Fenton reaction byproduct, confirms ˙OH generation and which could support the occurrence of intracellular Fenton chemistry (Fig. S36 and S37). These results demonstrate that TPPI functions as an O2˙− photogenerator capable of inducing oxidative stress under normoxic conditions and, importantly, that it could be effective even under hypoxic environments (≤2% O2) due to the partial O2-recycling pathway, thereby ensuring sustained photodynamic cytotoxicity toward cancer cells.
| H2O2 + Fe2+ → Fe3+ + OH˙ + OH− |
| | | O2˙− + Fe3+ → Fe2+ + 3O2 | (3) |
| | | O2˙− + H2O2 → 3O2 + OH˙ + OH− | (4) |
Conclusion
This study reports the careful tuning of the donor functionality in heavy-metal free PSs, with minor changes at the peri-position of the planar PI core, that yielded far-red/NIR (AIE/AIEE), significant Stokes shifts (213–270 nm), and exceptional (NIR AIEE/NIR) photoredox catalytic properties. The PSs DMPI/BPPI showed NIR/far-red AIEE photoredox catalytic properties for O2˙− generation. TPPI showed far-red AIE/NIR dual-type PS/photoredox (1O2/O2˙−) catalytic features containing ΦΔ of 0.59 in aqueous media, whereas PhPI/ANPI exhibited ACQ properties. ANPI functions as a photoredox catalyst for ˙OH/O2˙− (type-I PS), comprising exceptionally low ΔGOH (−413.93 kcal mol−1), whereas PhPI was a photoredox catalyst prominently for O2˙−. Notably, this study also showed that the dimethyl-induced NIR AIEE in the planar PI core features very prominently among the RPIs which results from the steric crowding around the pendant phenyl unit, which converts DMPI from ACQ into AIEE form. Distinct optical properties include DMPI: NIR AIEE (λem,max = 720 nm, Stokes shift = 220 nm) and NIR solid state emission (λem,max = 770 nm, Stokes shift = 270 nm), TPPI: far-red AIE (λem,max = 686 nm, Stokes shift = 175 nm) and NIR solid state emission (λem,max = 713 nm, Stokes shift = 213 nm), and BPPI: far-red AIEE (λem,max = 675 nm, Stokes shift = 186 nm) and NIR solid state emission (λem,max = 716 nm, Stokes shift = 216 nm). The RPIs are engineered strategically by introducing specific electron donor subunits to reduce ΔES0–T1, thereby hindering the efficient conversion of 3O2 to 1O2 that enabled efficient NIR photoredox catalytic properties. Consequently, this design principle has helped to fabricate distinct heavy-metal free (NIR AIEE/NIR) photoredox catalytic mechanisms that precisely generate O2˙−via the refined ΔES0–T1 principle. TPPI was engineered via triplet-ground-state splitting energy modulation functions through a partial O2-recycling pathway that induces significant cancel cell death, where O2˙− undergoes Haber–Weiss and Fenton reactions to amplify ROS generation.
Author contributions
MNK and PKI designed the experiments. MNK synthesized the materials and conducted all the characterization studies. MNK and PKI wrote and thoroughly revised the manuscript. SN carried out the in vitro cellular studies and fluorescence microscopic analysis. SN and SK analysed the cellular data. RDA, CS and MS performed the FESEM, TRPL and LC-MS experiments. All authors discussed the results and contributed to the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: materials, instruments, synthetic procedures, and characterization data (multinuclear NMR and MALDI-TOF), as well as experimental and computational data (Schemes S1–S3, Fig. S1–S37, Tables S1–S32, and SI figures). See DOI: https://doi.org/10.1039/d5sc07763j.
Acknowledgements
The authors gratefully acknowledge financial support from the Department of Electronics & Information Technology (DeitY Project No. 5(9)/2012-NANO Vol. II), the Department of Science and Technology (DST No. DST/SERB/EMR/2014/000034), and the DST-Max Planck Society, Germany (No. IGSTC/MPG/PG(PKI)/2011A/48). Gratitude is also extended to the Centre for Nanotechnology and the Central Instruments Facility at IIT Guwahati for instrument support, and Prof. Aditya Narayan Panda for assistance with theoretical calculations.
References
- Y. Shen, A. J. Shuhendler, D. Ye, J.-J. Xu and H.-Y. Chen, Chem. Soc. Rev., 2016, 45, 6725–6741 Search PubMed.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 Search PubMed.
- D. Cui, J. Huang, X. Zhen, J. Li, Y. Jiang and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 5920–5924 Search PubMed.
- J. Li, Z. Zhuang, Z. Zhao and B. Z. Tang, View, 2022, 3, 20200121 Search PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 Search PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- L. Benov, Protoplasma, 2001, 217, 33–36 Search PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun and K. Shao, J. Am. Chem. Soc., 2019, 141, 2695–2702 Search PubMed.
- M. Li, Y. Shao, J. H. Kim, Z. Pu, X. Zhao, H. Huang, T. Xiong, Y. Kang, G. Li and K. Shao, J. Am. Chem. Soc., 2020, 142, 5380–5388 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- A. Jana, L. Bai, X. Li, H. Ågren and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 2336–2347 CrossRef CAS PubMed.
- W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C. J. Zhang, X. Cheng, Y. Yuan, D. Ding and D. Kong, Adv. Mater., 2017, 29, 1700548 Search PubMed.
- R. Docampo, S. N. Moreno, R. P. Muniz, F. S. Cruz and R. P. Mason, Science, 1983, 220, 1292–1295 CrossRef CAS PubMed.
- M. N. Khatun, S. Nandy, C. Srinivas, S. Kumar and P. K. Iyer, Adv. Opt. Mater., 2025, 13, e01052 CrossRef CAS.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- M. N. Khatun, S. Nandy, H. Roy, S. S. Ghosh, S. Kumar and P. K. Iyer, Chem. Sci., 2024, 15, 9298–9317 Search PubMed.
- K. Chen, R. Zhang, Z. Wang, W. Zhang and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 1901433 CrossRef CAS.
- M. Jiang, X. Gu, R. T. Kwok, Y. Li, H. H. Sung, X. Zheng, Y. Zhang, J. W. Lam, I. D. Williams and X. Huang, Adv. Funct. Mater., 2018, 28, 1704589 CrossRef.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 Search PubMed.
- T.-y. Li, J. Benduhn, Z. Qiao, Y. Liu, Y. Li, R. Shivhare, F. Jaiser, P. Wang, J. Ma and O. Zeika, J. Phys. Chem. Lett., 2019, 10, 2684–2691 CrossRef CAS PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- A. P. Deshmukh, N. Geue, N. C. Bradbury, T. L. Atallah, C. Chuang, M. Pengshung, J. Cao, E. M. Sletten, D. Neuhauser and J. R. Caram, Chem. Phys. Rev., 2022, 3, 021401 CrossRef CAS.
- M. Más-Montoya and R. A. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 Search PubMed.
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS.
- D. Ding, C. C. Goh, G. Feng, Z. Zhao, J. Liu, R. Liu, N. Tomczak, J. Geng, B. Z. Tang and L. G. Ng, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed.
- Z. Yang, W. Qin, J. W. Lam, S. Chen, H. H. Sung, I. D. Williams and B. Z. Tang, Chem. Sci., 2013, 4, 3725–3730 RSC.
- W. Qin, K. Li, G. Feng, M. Li, Z. Yang, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 635–643 Search PubMed.
- J. Wang, C. Li, Q. Chen, H. Li, L. Zhou, X. Jiang, M. Shi, P. Zhang, G. Jiang and B. Z. Tang, Anal. Chem., 2019, 91, 9388–9392 Search PubMed.
- L. Meng, S. Jiang, M. Song, F. Yan, W. Zhang, B. Xu and W. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 26842–26851 CrossRef CAS PubMed.
- M. Ojha, M. Banerjee, M. Mandal, T. Singha, S. Ray, P. K. Datta, M. Mandal, A. Anoop and N. P. Singh, ACS Appl. Mater. Interfaces, 2024, 16, 21486–21497 CrossRef CAS PubMed.
- N. Meher and P. K. Iyer, Nanoscale, 2017, 9, 7674–7685 RSC.
- N. Meher and P. K. Iyer, Nanoscale, 2019, 11, 13233–13242 RSC.
- C.-C. Wu, E. Y. Li and P.-T. Chou, Chem. Sci., 2022, 13, 7181–7189 RSC.
- S. Fatayer, B. Schuler, W. Steurer, I. Scivetti, J. Repp, L. Gross, M. Persson and G. Meyer, Nat. Nanotechnol., 2018, 13, 376–380 CrossRef CAS PubMed.
- T. Zhang, J. Zhang, F. B. Wang, H. Cao, D. Zhu, X. Chen, C. Xu, X. Yang, W. Huang and Z. Wang, Adv. Funct. Mater., 2022, 32, 2110526 CrossRef CAS.
- N. Meher, M. N. Khatun, R. Parui and P. K. Iyer, Nanoscale, 2025, 17, 6685–6694 RSC.
- E. Bodunov and A. Simões Gamboa, J. Phys. Chem. C, 2018, 122, 10637–10642 CrossRef CAS.
- J. R. Martins, V. Krivenkov, C. R. Bernardo, P. Samokhvalov, I. Nabiev, Y. P. Rakovich and M. I. Vasilevskiy, J. Phys. Chem. C, 2022, 126, 20480–20490 CrossRef CAS PubMed.
- C. Liu, A. Rastogi and H.-C. Yeh, Anal. Chem., 2017, 89, 4772–4775 Search PubMed.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2009, 131, 662–670 CrossRef CAS PubMed.
- N. Meher, S. Panda, S. Kumar and P. K. Iyer, Chem. Sci., 2018, 9, 3978–3985 RSC.
- N. Meher, A. P. Bidkar, D. Barman, S. S. Ghosh and P. K. Iyer, Chem. Commun., 2020, 56, 14861–14864 Search PubMed.
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed.
- Z. Yang, Z. Zhang, Z. Lei, D. Wang, H. Ma and B. Z. Tang, ACS Nano, 2021, 15, 7328–7339 CrossRef CAS PubMed.
- L. Yu, Y. Xu, Z. Pu, H. Kang, M. Li, J. L. Sessler and J. S. Kim, J. Am. Chem. Soc., 2022, 144, 11326–11337 CrossRef CAS PubMed.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- K. X. Teng, W. K. Chen, L. Y. Niu, W. H. Fang, G. Cui and Q. Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 19912–19920 CrossRef CAS PubMed.
- H. Bian, D. Ma, X. Zhang, K. Xin, Y. Yang, X. Peng and Y. Xiao, Small, 2021, 17, 2100398 CrossRef CAS PubMed.
- P. Xiao, Z. Shen, D. Wang, Y. Pan, Y. Li, J. Gong, L. Wang, D. Wang and B. Z. Tang, Adv. Sci., 2022, 9, 2104079 CrossRef CAS PubMed.
- M. C. Catapano, M. Protti, T. Fontana, R. Mandrioli, P. Mladěnka and L. Mercolini, Molecules, 2019, 24, 3066 CrossRef CAS PubMed.
- A. Rahal, A. Kumar, V. Singh, B. Yadav, R. Tiwari, S. Chakraborty and K. Dhama, BioMed Res. Int., 2014, 2014, 761264 Search PubMed.
- B. Giasson, J. Duda, I. Murray, Q. Chen, J. M. Souza, H. I. Hurtig, H. Ischiropoulos, J. Q. Trojanowski and V. M. Lee, Science, 2000, 290, 985–989 CrossRef CAS PubMed.
- T. M. Dawson and V. L. Dawson, Science, 2003, 302, 819–822 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Satyendu
Nandy
a,
Satyendu
Nandy









