
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organo-catalyzed photoelectrochemical ring-contraction of arylidenecyclobutanols via radical cation-triggered semipinacol rearrangement
Yu
Zheng
 *,
Chunxi
Chen
,
Xuhao
Zhou
,
Guoyang
Deng
,
Yanju
Lu
and
Shenlin
Huang
*
*,
Chunxi
Chen
,
Xuhao
Zhou
,
Guoyang
Deng
,
Yanju
Lu
and
Shenlin
Huang
*
State Key Laboratory for Development and Utilization of Forest Food Resources, Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, China. E-mail: zhengy@njfu.edu.cn; shuang@njfu.edu.cn
First published on 28th November 2025
Abstract
The semipinacol rearrangement has proven to be an efficient strategy for converting allylic alcohols into carbonyl compounds bearing an α-quaternary carbon center. Traditionally, this semipinacol rearrangement is initiated through two primary modes, namely electrophile and radical-triggered pathways. Notably, these two strategies predominantly lead to ring-expansion β-functionalized products from cyclic allylic alcohols. In contrast, the radical cation, possessing carbon-centered radical and electrophilic carbocation dual reactivity, potentially triggers ring contraction semipinacol rearrangement of cyclic allylic alcohols, but remains underexplored. This is presumably due to the favorable C–C bond cleavage facilitated by ring-strain release and the challenge associated with overcoming the energy barriers required for ring-contraction. Herein, we report the first photoelectrochemical alkene radical cation-triggered semipinacol rearrangement for the ring-contraction of arylidenecyclobutanols. This methodology enables access to diverse and valuable 1,1-cyclopropane formylketones and diketones under mild and environmentally benign conditions. The resulting products not only contain the cyclopropane motif, which is present in many pharmaceuticals and bioactive molecules, but also serve as useful synthetic intermediates for the preparation of various derivatives, including the key intermediate of cabozantinib.
Introduction
Rearrangement reactions represent powerful tools for skeletal remodeling. These reactions fundamentally alter the framework by cleaving existing bonds and reorganizing atoms or groups within the molecule. Crucially, rearrangement reactions enable the efficient construction of complex and often difficult-to-access carbon skeletons directly from simple precursors, bypassing multi-step synthetic routes. This capability is particularly valuable in the synthesis of intricate natural products and pharmaceuticals.1–4 Among various rearrangement reactions, the semipinacol rearrangement has proven to be an efficient strategy for the transformation of allylic alcohols to carbonyl compounds with an α-quaternary carbon center.5–7 Traditionally, the electrophilic attack toward the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of allylic alcohol generates an electrophilic carbocation, which triggers the subsequent rearrangement, delivering the corresponding carbonyl compounds (Fig. 1A).8–18 An alternative strategy involves the radical addition of the CC bond to form a carbon-centered radical, which is further oxidized to a carbocation intermediate (radical-polar crossover process), thereby triggering semipinacol rearrangement. This strategy has recently emerged as an efficient platform for the reaction of various radical precursors with allylic alcohols to access diverse β-functionalized ketones via photocatalysis19–23 or electrocatalysis,24–27 among others.28–31 Furthermore, both electrophilic and radical functionalization/semipinacol rearrangement sequences typically yield ring-expansion β-functionalized products from cyclic allylic alcohols. This is because the β-C−C bond cleavage of cyclic allylic alcohols is more favored than other transformations due to the release of ring strain. For example, the electrophilic fluorination of arylidenecyclobutanols generated a carbocation intermediate, which tended to undergo C–C bond cleavage, thereby delivering the ring-opening products disclosed by Zhao's group in 2017 (Fig. 1B).32 More recently, the Ackermann,33 Zhang,34 and our groups35 independently reported that the photocatalytic radical addition of arylidenecyclobutanols led to the same C–C bond cleavage driven by strain release, resulting in the ring-opening products. On the other hand, given the strain energies of 26.3 kcal mol−1 for cyclobutanes versus 29.0 kcal mol−1 for cyclopropanes,36 the ring-contraction of cyclobutanols to access cyclopropanes should be thermodynamically challenging. In 2019, Frongia et al. reported the synthesis of cyclopropanecarbaldehydes through a tandem Wittig reaction and ring-contraction process at 80 °C over 2 days (Fig. 1C).37 Separately, Li and coworkers accomplished a fluorination/semipinacol rearrangement cascade for the ring-contraction of 2-alkylidenecyclobutanol, using a combination of nucleophilic Py·HF and (PhIO)n.38 Furthermore, alkoxy radical mediated β-C−C bond cleavage of cyclobutanols generates carbon-centered radicals capable of participating in diverse transformations.39–47 This competing reaction pathway thereby jeopardizes the desired ring-contraction process. To the best of our knowledge, the radical cation, which features carbon-centered radical and electrophilic carbocation dual reactivity and directly triggers the ring-contraction semipinacol rearrangement of cyclic allylic alcohols, remains underexplored.
C bond of allylic alcohol generates an electrophilic carbocation, which triggers the subsequent rearrangement, delivering the corresponding carbonyl compounds (Fig. 1A).8–18 An alternative strategy involves the radical addition of the CC bond to form a carbon-centered radical, which is further oxidized to a carbocation intermediate (radical-polar crossover process), thereby triggering semipinacol rearrangement. This strategy has recently emerged as an efficient platform for the reaction of various radical precursors with allylic alcohols to access diverse β-functionalized ketones via photocatalysis19–23 or electrocatalysis,24–27 among others.28–31 Furthermore, both electrophilic and radical functionalization/semipinacol rearrangement sequences typically yield ring-expansion β-functionalized products from cyclic allylic alcohols. This is because the β-C−C bond cleavage of cyclic allylic alcohols is more favored than other transformations due to the release of ring strain. For example, the electrophilic fluorination of arylidenecyclobutanols generated a carbocation intermediate, which tended to undergo C–C bond cleavage, thereby delivering the ring-opening products disclosed by Zhao's group in 2017 (Fig. 1B).32 More recently, the Ackermann,33 Zhang,34 and our groups35 independently reported that the photocatalytic radical addition of arylidenecyclobutanols led to the same C–C bond cleavage driven by strain release, resulting in the ring-opening products. On the other hand, given the strain energies of 26.3 kcal mol−1 for cyclobutanes versus 29.0 kcal mol−1 for cyclopropanes,36 the ring-contraction of cyclobutanols to access cyclopropanes should be thermodynamically challenging. In 2019, Frongia et al. reported the synthesis of cyclopropanecarbaldehydes through a tandem Wittig reaction and ring-contraction process at 80 °C over 2 days (Fig. 1C).37 Separately, Li and coworkers accomplished a fluorination/semipinacol rearrangement cascade for the ring-contraction of 2-alkylidenecyclobutanol, using a combination of nucleophilic Py·HF and (PhIO)n.38 Furthermore, alkoxy radical mediated β-C−C bond cleavage of cyclobutanols generates carbon-centered radicals capable of participating in diverse transformations.39–47 This competing reaction pathway thereby jeopardizes the desired ring-contraction process. To the best of our knowledge, the radical cation, which features carbon-centered radical and electrophilic carbocation dual reactivity and directly triggers the ring-contraction semipinacol rearrangement of cyclic allylic alcohols, remains underexplored.
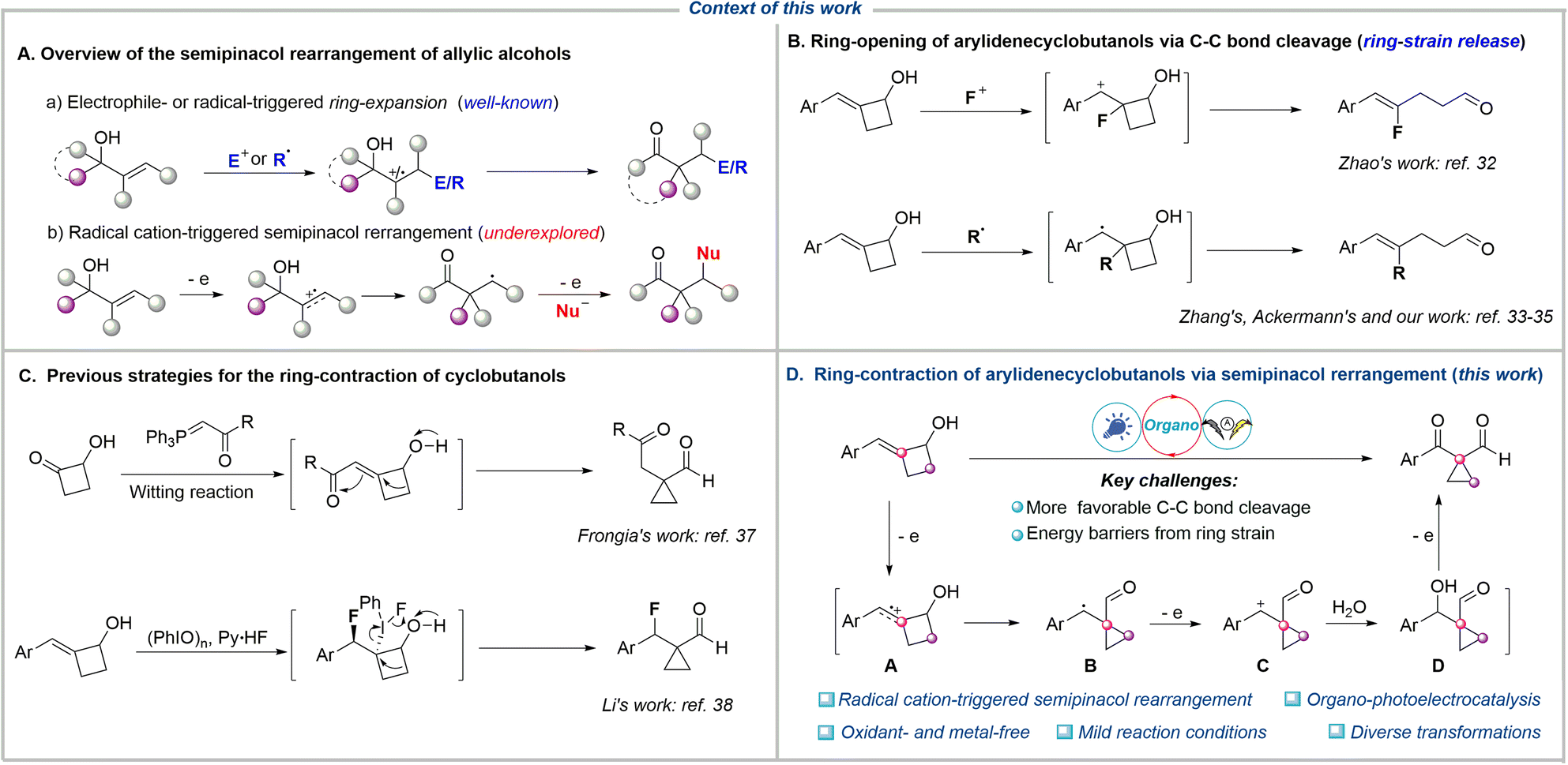 | ||
| Fig. 1 Context of this work. (A) Overview of the semipinacol rearrangement of allylic alcohols. (B) Ring-opening of arylidenecyclobutanols via C–C bond cleavage. (C) Previous strategies for the ring-contraction of cyclobutanols. (D) Ring-contraction of arylidenecyclobutanols via semipinacol rearrangement (this work). | ||
Photoelectrochemistry leverages the synergistic integration of light and electrical energy to achieve challenging reactivity and selectivity under mild and environmentally benign conditions.48–55 This approach significantly broadens the accessible redox window for reaction design while eliminating dependence on chemical redox reagents, establishing a new paradigm for green and precision organic synthesis. Based on recent achievements in alkene radical cation chemistry56–62 and combined with our continuous interest in electrochemical synthesis of cyclopropanes,63 we envisioned that chemoselective oxidation of the CC bonds in arylidenecyclobutanols could generate the corresponding alkene radical cation A, which possesses carbon-centered radical and electrophilic carbocation dual reactivity, and thereby might trigger the semipinacol rearrangement to give the ring-contraction intermediate B under suitable conditions (Fig. 1D). Subsequent oxidation of intermediate B followed by nucleophilic addition results in the final cyclopropanes, which are important motifs present in many pharmaceuticals and bioactive molecules.64–70 Herein, we report an organo-catalyzed photoelectrochemical ring-contraction of cyclobutanols via alkene radical cation triggered-semipinacol rearrangement to access diverse and valuable 1,1-cyclopropane formylketones and diketones with an α-quaternary carbon center under environmentally friendly and mild reaction conditions without the requirement of an oxidant and metal-catalyst.
Results and discussion
The feasibility of our proposal was investigated by employing 2-benzylidenecyclobutanol 1a as the model substrate under photoelectrochemical conditions (Table 1). After systematically optimizing different reaction parameters, the desired ring-contraction product 1-benzoyl-1-formylcyclopropane 2a was finally obtained in 72% isolated yield under the following optimal conditions: the photoelectrolysis of 1a was performed in the presence of 5 mol% 9-mesityl-10-methyl acridinium perchlorate ([Mes-Acr+]ClO4−) as the catalyst, nBu4NPF6 as an electrolyte, Pt plates as the electrode materials in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 CH3CN/H2O mixed solvent at a constant current of 5 mA and 40 W blue light irradiation for 6 hours (Table 1, entry 1). The solvent effect had a decisive influence, as no desired product was detected when using other solvents such as dimethylformamide (DMF) or tetrahydrofuran (THF) instead of CH3CN (Table 1, entry 2, for more details see SI, Table S1). The electrode materials had a great impact on the reaction efficiency. Switching the cathode material from Pt(−) to Sn(−) or C(−) afforded a decreased yield, whereas on using GF(+) or C(+) as the anode, only a trace amount of the product was observed (Table 1, entries 3 and 4). Among the tested catalysts, including organic dyes, metal photocatalysts, and modifications of [Mes-Acr+]ClO4−, no better results were obtained (Table 1, entries 5 and 6, for more details see SI, Table S1). Performing the photoelectrochemical reaction with 15 W and 35 W blue LEDs resulted in a diminished yield in both cases (Table 1, entry 7). Slight variations in decreasing or increasing the amount of H2O had a deleterious influence on the outcome (Table 1, entry 8). The replacement of nBu4NPF6 with other electrolytes such as Et4NPF6 or NH4PF6 led to reduced yields (Table 1, entry 9, for more details see SI, Table S1). In addition, either decreasing or increasing the constant current provided a lower yield (Table 1, entry 10). Furthermore, the addition of trifluoroacetic acid (TFA) or base (K2CO3) led to no improvement in the yield (Table 1, entry 11). Control experiments indicated that without light or a catalyst, the yield dropped to 31% or 34%, respectively (Table 1, entries 12 and 13). In addition, electricity is crucial to the reaction since the yield was significantly decreased to 12% without the constant current (Table 1, entry 14). These results indicated that electricity, light as well as the catalyst together guarantee the reaction efficiency. Furthermore, the observation of α,β-unsaturated cyclobutanone and 1-benzoylcyclopropane-1-carboxylic acid as the main byproducts under optimal conditions accounted for the relatively low yield of 2a (see the SI for more details).
:1.2 CH3CN/H2O mixed solvent at a constant current of 5 mA and 40 W blue light irradiation for 6 hours (Table 1, entry 1). The solvent effect had a decisive influence, as no desired product was detected when using other solvents such as dimethylformamide (DMF) or tetrahydrofuran (THF) instead of CH3CN (Table 1, entry 2, for more details see SI, Table S1). The electrode materials had a great impact on the reaction efficiency. Switching the cathode material from Pt(−) to Sn(−) or C(−) afforded a decreased yield, whereas on using GF(+) or C(+) as the anode, only a trace amount of the product was observed (Table 1, entries 3 and 4). Among the tested catalysts, including organic dyes, metal photocatalysts, and modifications of [Mes-Acr+]ClO4−, no better results were obtained (Table 1, entries 5 and 6, for more details see SI, Table S1). Performing the photoelectrochemical reaction with 15 W and 35 W blue LEDs resulted in a diminished yield in both cases (Table 1, entry 7). Slight variations in decreasing or increasing the amount of H2O had a deleterious influence on the outcome (Table 1, entry 8). The replacement of nBu4NPF6 with other electrolytes such as Et4NPF6 or NH4PF6 led to reduced yields (Table 1, entry 9, for more details see SI, Table S1). In addition, either decreasing or increasing the constant current provided a lower yield (Table 1, entry 10). Furthermore, the addition of trifluoroacetic acid (TFA) or base (K2CO3) led to no improvement in the yield (Table 1, entry 11). Control experiments indicated that without light or a catalyst, the yield dropped to 31% or 34%, respectively (Table 1, entries 12 and 13). In addition, electricity is crucial to the reaction since the yield was significantly decreased to 12% without the constant current (Table 1, entry 14). These results indicated that electricity, light as well as the catalyst together guarantee the reaction efficiency. Furthermore, the observation of α,β-unsaturated cyclobutanone and 1-benzoylcyclopropane-1-carboxylic acid as the main byproducts under optimal conditions accounted for the relatively low yield of 2a (see the SI for more details).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yielda of 2a [%] |
| a GC yields with 1-nitronaphthalene as the internal standard. b Isolated yield. c n.d. = No desired product was detected. | ||
| 1 | None | 59 (72)b |
| 2 | THF or DMF instead of CH3CN | n.d.c |
| 3 | C(−) or Sn(−) instead of Pt(−) | 35, 32 |
| 4 | C(+) or GF(+) instead of Pt(+) | Trace, trace |
| 5 | 4CzIPN instead of [Mes-Acr+]ClO4− | 48 |
| 6 | Ir(dF(CF3)ppy)2(dtbbpy)]PF6 instead | 52 |
| 7 | 15 W or 35 W instead of 40 W | 41, 54 |
| 8 | 1.0 mL or 1.5 mL H2O instead | 34, 50 |
| 9 | Et4NPF6 or NH4PF6 instead | 42, 58 |
| 10 | 3 mA or 8 mA instead of 5 mA | 54, 37 |
| 11 | TFA or K2CO3 as additives | 49, 35 |
| 12 | No light | 31 |
| 13 | No catalyst | 34 |
| 14 | No electricity | 12 |
With the optimal conditions in hand, we moved our attention to the substrate scope of this photoelectrochemical ring-contraction reaction, as illustrated in Fig. 2. First, we investigated the modifications of the substituents at the para-position of the aryl ring of arylidenecyclobutanols. In general, both electron-donating (–Me, –tBu, –OMe, and –Ph) and electron-withdrawing groups (–F, –Cl, –Br, and –CO2Et) all tolerated very well, delivering the corresponding cyclopropanes 2b–2i in good yields. Subsequently, the compatibility of functional groups at the meta and ortho positions of the aryl ring was evaluated. Both electron-donating (–Me) and electron-withdrawing (–Br) substituents reacted smoothly, affording the desired cyclopropanes 2j–2m in moderate to good yields. In addition, sensitive functional group alkyne was well tolerated (2n). Diverse di-substituted derivatives were applicable to this catalytic reaction, and the desired products (2o–2s) were isolated in moderate to good yields. Arylidenecyclobutanols bearing a naphthyl ring, benzothiophene, and thiophene were competent substrates, giving rise to the desired cyclopropanes 2t–2v. Apart from the above secondary alcohols, tertiary alcohol substrates were subsequently accessed under otherwise identical conditions. Methyl- and isopropyl-substituted arylidenecyclobutanols 1w and 1x were smoothly transformed into 2w and 2x in 51% and 58% yields, respectively. Moreover, phenyl-substituted arylidenecyclobutanol 1y exhibited good reactivity in this photoelectrochemical ring-contraction reaction as well. Arylidenecyclobutanol with a dimethyl group on the four-member ring was suitable under the standard conditions, delivering the cyclopropane derivatives in 43% yield (2z). Notably, the photoelectrochemical conditions allowed us to explore substrates derived from more complex molecules, accessing the target cyclopropanes with L-menthol, (+)-isopulegol, and (−)-borneol (2aa–2ac). It is noteworthy that the relatively low yields were mainly attributable to the formation of oxidation byproducts that were also observed in the model reaction. Meanwhile, the reaction suffers from some limitations. The cyclohexyl-substituted cyclobutanol substrate was not well-tolerated, likely due to the instability of the alkyl radical intermediate. Benzylidenecyclopentanol was unsuccessful under the standard conditions. When the substrate containing an oxetane motif was applied to the reaction, the corresponding product was not observed, with the starting materials being completely consumed, indicating that the oxygen atom migration failed under current conditions.
 | ||
| Fig. 2 Substrate scope investigations. (A) Substrate scope. (B) Unsuccessful examples. | ||
To obtain experimental evidence of the reaction mechanism, we conducted a series of experiments (Fig. 3A). The addition of radical scavengers, including 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and butylated hydroxytoluene (BHT), both sharply suppressed the formation of the desired product (Fig. 3B). On conducting the reaction with H218O instead of H2O under standard conditions, the heavy-oxygen-labelled product 2a-18O was formed, which was confirmed by high-resolution mass spectrometry (HRMS), suggesting that oxygen comes from water and water serves as the nucleophile in the reaction (Fig. 3C). Since substrate 1a contains a hydroxyl group, which could generate the alkoxy radical under oxidation conditions. This reactive radical can undergo β-scission, radical addition, oxidation, and nucleophilic attack sequences, eventually affording the same product. To rule out this possibility, trimethylsilyl alkenylcyclobutanol 3 was subjected to the reaction, and product 2a was isolated in 45% yield, indicating that the alkoxy radical was not involved in the reaction (Fig. 3D). In addition, cyclic voltammetry was employed to identify the oxidation sequence of the alkene and hydroxyl group. Substrate 1a shows two oxidation peaks at E = 1.36 and 2.02 V vs. Ag/AgCl, whereas benzylidenecyclobutane 4 presents an oxidation peak at 1.29 V vs. Ag/AgCl, demonstrating the preferential oxidation of the alkene bond of 1 (Fig. 3E). Furthermore, the luminescence quenching experiments were performed with 1a and [Mes-Acr+]ClO4− (Fig. 3F). The results showed that the excited state of [Mes-Acr+]ClO4− was quenched by 1a, signifying that 1a was oxidized by the excited photocatalyst (see the SI for more details).
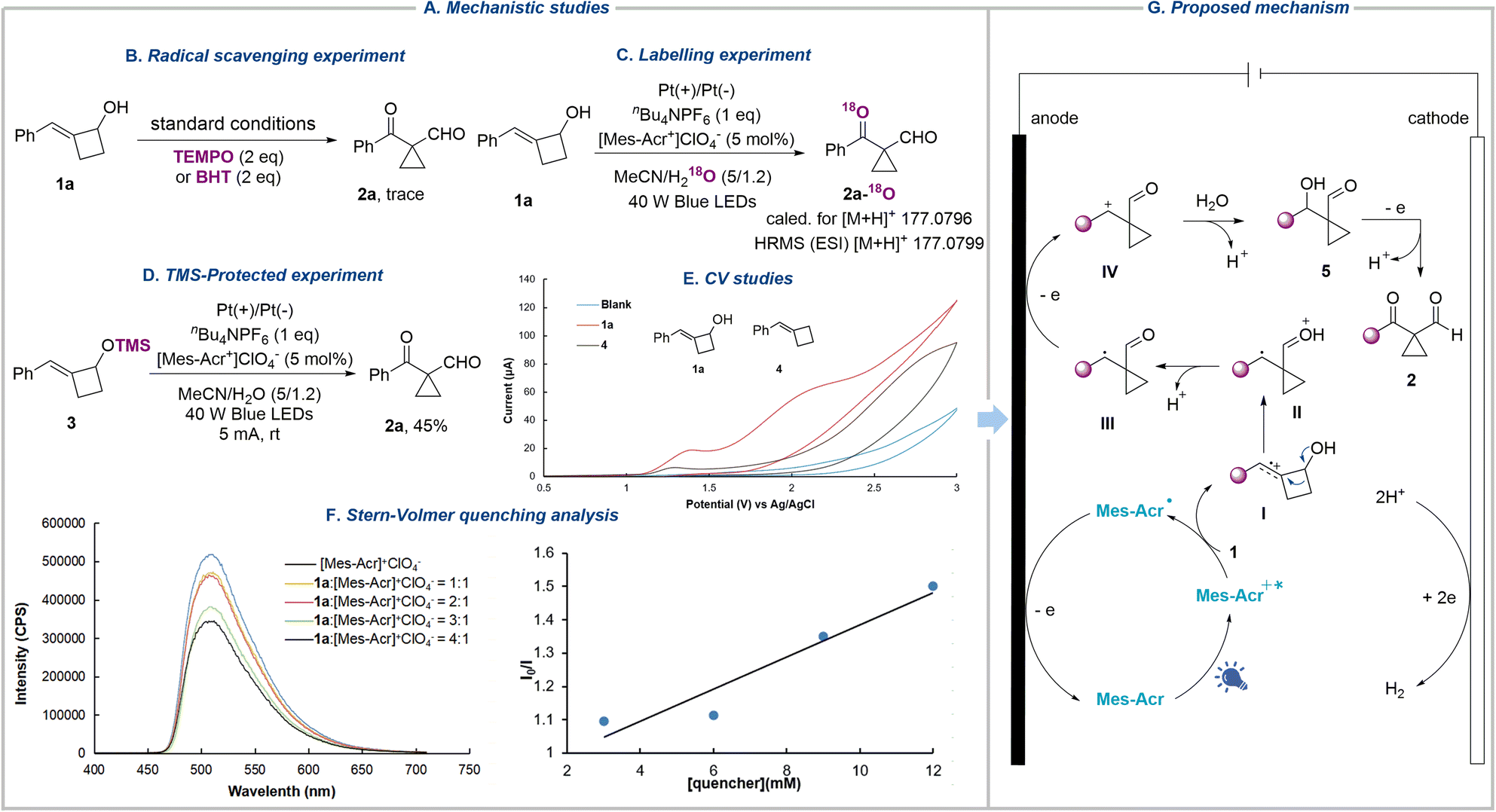 | ||
| Fig. 3 Mechanistic studies and proposal. (A) Mechanistic studies. (B) Radical scavenging experiment. (C) Labelling experiment. (D) TMS-protected experiment. (E) CV studies. (F) Stern–Volmer quenching analysis. (G) Proposed mechanism. | ||
Based on the above data and previous work,71–73 we propose the mechanism of this photoelectrochemically driven semipinacol-type rearrangement reaction as shown in Fig. 3G. Initially, the excited-state organic dye photocatalyst Mes-Acr+* (Ered = 2.06 V vs. SCE in MeCN)74 could sufficiently oxidize the double bond of 1 to generate the radical cation I and the acridinyl radical Mes-Acr˙, which is oxidized at the anode to regenerate the ground-state organocatalyst Mes-Acr+. The radical cation I, featuring carbon-centered radical and electrophilic carbocation dual reactivity, undergoes semipinacol-type rearrangement to give the oxonium ion intermediate II, which then transforms into the carbon-centered radical III after the loss of a proton. Subsequently, radical III is further oxidized to give the cation intermediate IV, which is attacked by H2O acting as a nucleophile, resulting in the formation of 5 followed by deprotonation. Compound 5 was observed during the reaction, which disappeared after the completion of the reaction (see the SI for more details). Finally, the third anodic oxidation delivers cyclopropanes 2.
As illustrated in Fig. 4A, further transformations of these 1,1-disubstituted cyclopropanes were investigated to highlight their synthetic utility. First, the formyl group in 2a could be chemoselectively reduced by NaBH4 to give 6 in 60% yield. Using a stronger reductant, LiAlH4, the carbonyl group was also reduced, leading to the formation of diol 7 in 78% yield. On the other hand, the oxidation of 2a proceeded smoothly and afforded the acid 8, whose structure was determined by X-ray crystallographic analysis. The reaction of compound 2a with allyl bromide in the presence of Zn successfully gave rise to allylic alcohol 9. Moreover, the condensation reaction of 2a between TsNHNH2 yielded the corresponding tosylhydrazone 10 in 74% yield. Finally, the oxidation of 2f with Oxone afforded compound 11 in 75% yield (Fig. 4B). The subsequent Beckmann rearrangement successfully delivered compound 12, a key intermediate toward the synthesis of cabozantinib.75 This alternative synthetic route demonstrated the practicality of this methodology, featuring an advantage that eliminates the use of corrosive and toxic SOCl2.
 | ||
| Fig. 4 Synthetic applications. (A) Product derivatizations. (B) Synthesis of cabozantinib's key intermediate 12. | ||
Conclusions
In conclusion, we have developed the photoelectrochemical ring-contraction of arylidenecyclobutanols for the first time. This strategy provides a mild and environmentally benign route to a wide range of valuable 1,1-cyclopropane formylketones and diketones bearing an α-quaternary carbon center. Mechanistic investigations indicated that the alkene radical cation-triggered semipinacol rearrangements, deprotonation, oxidation, and nucleophilic attack sequences enable the desired cyclopropanes. The obtained products could serve as versatile synthetic intermediates, as demonstrated by their application in synthesizing a key intermediate of cabozantinib.Author contributions
Y. Z. and S. H. contributed to the conception and design of the experiments. Y. Z. directed the project and analyzed the data. C. C., X. Z., and G. D. performed the experiments and analyzed the data. Y. Z. wrote the manuscript. Y. L. and S. H. provided valuable suggestions to the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2455791 (8) contains the supplementary crystallographic data for this paper.76The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d5sc07637d.
Acknowledgements
Financial support provided by the National Natural Science Foundation of China (22201133, 32171724, and 32271818) is gratefully acknowledged. We also acknowledge financial support from the Natural Science Foundation of Jiangsu Province (BK20210607).Notes and references
- E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133–3148 RSC.
- R. A. Fernandes, P. Kattanguru, S. P. Gholapa and D. A. Chaudharia, Org. Biomol. Chem., 2017, 15, 2672–2710 RSC.
- X.-L. Liu, M.-L. Gong, X. Yang, P. Tian and Q.-H. Li, Org. Chem. Front., 2024, 11, 4934–4953 RSC.
- L. Chen, G. Li and L. Zu, Org. Chem. Front., 2022, 9, 5383–5394 RSC.
- S. K. Nanda, Adv. Synth. Catal., 2023, 365, 834–853 CrossRef.
- T. J. Snape, Chem. Soc. Rev., 2007, 36, 1823–1842 RSC.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed.
- Y. Xing, C. Li, J. P. Meng, Z. Zhang, X. Wang, Z. C. Wang, Y. Ye and K. Sun, Adv. Synth. Catal., 2021, 363, 3913–3936 CrossRef CAS.
- X.-M. Zhang, B.-S. Li, S.-H. Wang, K. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Sci., 2021, 12, 9262–9274 RSC.
- H. Lindner and E. M. Carreira, Angew. Chem., Int. Ed., 2024, 63, e202407827 CrossRef CAS.
- P. Zhao, W. Wang and T. Gulder, Org. Lett., 2023, 25, 6560–6565 CrossRef CAS.
- D.-Y. Zhang, Y. Zhang, H. Wu and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 7450–7453 CrossRef CAS.
- P. G. Kalomenopoulos, B. Emayavaramban and C. P. Johnston, Angew. Chem., Int. Ed., 2025, 64, e202414342 CrossRef CAS.
- M. A. S. Blackburn, C. C. Wagen, M. R. Bodrogean, P. M. Tadross, A. J. Bendelsmith, D. A. Kutateladze and E. N. Jacobsen, J. Am. Chem. Soc., 2023, 145, 15036–15042 CrossRef CAS.
- X. Yu, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2024, 146, 25878–25887 CrossRef CAS PubMed.
- S. Zhao, W. Yue, M. Yang, X. Li, B. Chen, Y. Gao, W. Yu, H.-L. Ni, P. Hu, B.-Q. Wang and P. Cao, Org. Lett., 2024, 26, 1224–1228 CrossRef CAS PubMed.
- T. Zheng, R. Chen, J. Huang, T. P. Gonçalves, K.-W. Huang and Y.-Y. Yeung, Chem, 2023, 9, 1255–1269 CAS.
- E. Capel, M. Rodríguez-Rodríguez, U. Uria, M. Pedron, T. Tejero, J. L. Vicario and P. Merino, J. Org. Chem., 2022, 87, 693–707 CrossRef CAS PubMed.
- S. Yao, K. Zhang, Q.-Q. Zhou, Y. Zhao, D.-Q. Shi and W.-J. Xiao, Chem. Commun., 2018, 54, 8096–8099 RSC.
- R. Giri, S. Patra and D. Katayev, ChemCatChem, 2023, 15, e202201427 CrossRef CAS.
- M. Liu, H. Huang and Y. Chen, Chin. J. Chem., 2018, 36, 1209–1212 CrossRef CAS.
- T.-C. Ma, S. Yao, M.-M. Qiao, F. Yuan, D.-Q. Shi and W.-J. Xiao, Org. Chem. Front., 2021, 8, 4224–4229 RSC.
- T. Kodo, K. Nagao and H. Ohmiya, Nat. Commun., 2022, 13, 2684 CrossRef CAS.
- S. J. Kwon and D. Y. Kim, Org. Lett., 2016, 18, 4562–4565 CrossRef CAS PubMed.
- H. I. Jung, Y. Kim and D. Y. Kim, Org. Biomol. Chem., 2019, 17, 3319–3323 RSC.
- J.-C. Kang, Y.-Q. Tu, J.-W. Dong, C. Chen, J. Zhou, T.-M. Ding, J.-T. Zai, Z.-M. Chen and S.-Y. Zhang, Org. Lett., 2019, 21, 2536–2540 CrossRef CAS PubMed.
- Y. J. Kim and D. Y. Kim, Org. Lett., 2019, 21, 1021–1025 CrossRef CAS PubMed.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Angew. Chem., Int. Ed., 2019, 58, 12491–12496 CrossRef CAS PubMed.
- S. Kayal, J. Kikuchi, M. Shimizu and M. Terada, ACS Catal., 2019, 9, 6846–6850 CrossRef CAS.
- B. Sahoo, J.-L. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577–11580 CrossRef CAS.
- V. Smyrnov and J. Waser, Org. Lett., 2023, 25, 6999–7003 CrossRef CAS PubMed.
- T.-L. Liu, J.-E. Wu and Y. Zhao, Chem. Sci., 2017, 8, 3885–3890 RSC.
- Y. Zhang, C. Zhao, C. Ma, Z. Cai, S. Trienes and L. Ackermann, Angew. Chem., Int. Ed., 2023, 62, e202300166 CrossRef CAS PubMed.
- C. Zhao, W. Ma, K. Liu, R. Xu, X. Ma and Y. Zhang, Org. Chem. Front., 2024, 11, 4663–4670 RSC.
- T. He, C. Liang, P. Jiang, H. Liang, S. Liao and S. Huang, Org. Lett., 2024, 26, 5577–5581 CrossRef CAS PubMed.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103–8112 CrossRef CAS.
- F. Cuccu, L. Serusi, A. Luridiana, F. Secci, P. Caboni, D. J. Aitken and A. Frongia, Org. Lett., 2019, 21, 7755–7758 CrossRef CAS.
- S.-X. Feng, S. Yang, F.-H. Tu, P.-P. Lin, L.-L. Huang, H. Wang, Z.-S. Huang and Q. Li, J. Org. Chem., 2021, 86, 6800–6812 CrossRef CAS.
- X. Wu, H. Zhang, N. Tang, Z. Wu, D. Wang, M. Ji, Y. Xu, M. Wang and C. Zhu, Nat. Commun., 2018, 9, 3343 CrossRef.
- D. Wang, J. Mao and C. Zhu, Chem. Sci., 2018, 9, 5805–5809 RSC.
- B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed.
- K. Zhao, K. Yamashita, J. E. Carpenter, T. C. Sherwood, W. R. Ewing, P. T. W. Cheng and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 8752–8757 CrossRef CAS.
- M. D. Hareram, A. A. M. A. EI Gehani, J. Harnedy, A. C. Seastram, A. C. Jones, M. Burns, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2022, 24, 3890–3895 CrossRef CAS PubMed.
- M. Ju, S. Lee, H. M. Marvich and S. Lin, J. Am. Chem. Soc., 2024, 146, 19696–19703 CrossRef CAS.
- Z. Yang, D. Yang, J. Zhang, C. Tan, J. Li, S. Wang, H. Zhang, Z. Huang and A. Lei, J. Am. Chem. Soc., 2022, 144, 13895–13902 CrossRef CAS PubMed.
- L. Zhao, P. Hu, J. Tian, X. Zhang, C. Yang, L. Guo and W. Xia, Org. Lett., 2024, 26, 4882–4886 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS.
- J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS.
- L. Qian and M. Shi, Chem. Commun., 2023, 59, 3487–3506 RSC.
- M. C. Lamb, K. A. Steiniger, L. K. Trigoura, J. Wu, G. Kundu, H. Huang and T. H. Lambert, Chem. Rev., 2024, 124, 12264–12304 CrossRef CAS PubMed.
- L. Li, Y. Yao and N. Fu, Chem Catal., 2024, 4, 100898 CAS.
- P. Xiong and H.-C. Xu, Acc. Chem. Res., 2025, 58, 299–311 CrossRef CAS PubMed.
- M.-J. Luo, Q. Xiao and J.-H. Li, Chem. Soc. Rev., 2022, 51, 7206–7237 RSC.
- N. Chen and H.-C. Xu, Chem. Rec., 2021, 21, 2306–2319 CrossRef CAS.
- P. Xiong, H. Long, J. Song, Y. Wang, J.-F. Li and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 16387–16391 CrossRef CAS.
- H. Huang and T. H. Lamber, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS.
- K. Rybicka-Jasińska, Z. Szeptuch, H. Kubiszewski and A. Kowaluk, Org. Lett., 2023, 25, 1142–1146 CrossRef PubMed.
- H. Yi, L. Niu, C. Song, Y. Li, B. Dou, A. K. Singh and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 1120–1124 CrossRef CAS PubMed.
- G. Zhang, X. Hu, C.-W. Chiang, H. Yi, P. Pei, A. K. Singh and A. Lei, J. Am. Chem. Soc., 2016, 138, 12037–12040 CrossRef CAS PubMed.
- W. Yi, P.-C. Xu, T. He, S. Shi and S. Huang, Nat. Commun., 2024, 15, 9645 CrossRef CAS.
- L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1648 CrossRef CAS.
- D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS.
- Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China Chem., 2016, 59, 1126–1141 CrossRef CAS.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS.
- J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Nat. Prod. Rep., 2022, 39, 926–945 RSC.
- H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- D. Chen, Y. Wang, Y. Ma, B. Xiong, J. Ai, Y. Chen, M. Geng and J. Shen, ChemMedChem, 2012, 7, 1057–1070 CrossRef CAS PubMed.
- C. Chen, CCDC 2455791: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nfg2n.
| This journal is © The Royal Society of Chemistry 2026 |