
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical Cu(III)-mediated trifluoromethylation of (hetero)arenes and biomolecules
Petr
Pospíšil
 ab,
Vladimir
Motornov
*ad,
Ondřej
Michal
ac,
Lucie
Šálková
ac,
Soňa
Boháčová
a,
Tomáš
Slanina
a,
Ján
Tarábek
a,
Blanka
Klepetářová
a and
Petr
Beier
*a
ab,
Vladimir
Motornov
*ad,
Ondřej
Michal
ac,
Lucie
Šálková
ac,
Soňa
Boháčová
a,
Tomáš
Slanina
a,
Ján
Tarábek
a,
Blanka
Klepetářová
a and
Petr
Beier
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 16600, Prague, Czech Republic. E-mail: beier@uochb.cas.cz; cuprate51@gmail.com
bInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210, Pardubice, Czech Republic
cDepartment of Physical Chemistry, University of Chemistry and Technology, Prague, Technická 5, 16628, Prague, Czech Republic
dFreie Universität Berlin, Fabeckstraße 34-36, 14195 Berlin, Germany
First published on 11th November 2025
Abstract
A highly efficient and atom-economical method for the C–H trifluoromethylation of (hetero)arenes and complex biomolecules has been developed using a substoichiometric amount of the stable tetrakis(trifluoromethyl)cuprate(III) salt. Upon violet-light irradiation in the presence of an oxidant, all four CF3 groups are sequentially converted into trifluoromethyl radicals, enabling high-yielding transformations under mild conditions. The protocol exhibits excellent functional group tolerance and is applicable to the late-stage trifluoromethylation of pharmaceuticals, amino acids, and nucleosides. Mechanistic studies support a photoinitiated radical pathway and reveal the full utilization of the Cu(III) species. The results presented advance the use of copper-mediated strategies for the sustainable incorporation of fluorine into complex molecules.
Introduction
Cu(III) trifluoromethyl compounds have received wide interest from organic and inorganic chemists due to their exceptional stability and versatile reactivity (Scheme 1A).1 Since the breakthrough discovery by Grushin in 2015, who described a simple procedure for the preparation of tetrakis(trifluoromethyl)cuprate salts [Cu(CF3)4]− from CuCl with air as the only oxidant,2 Cu(III) trifluoromethyl complexes have become candidates for applications in organic synthesis, especially as trifluoromethylation agents.1a However, homoleptic tetrakis(trifluoromethyl)-cuprate(III) salts are known to be poor trifluoromethylation agents. They have been shown to be activated to release trifluoromethyl radicals by harsh UV irradiation with only low efficiency (Scheme 1B),3 or by electrospray ionization.4 Therefore, neutral complexes with bidentate N-donor ligands, such as (bpy)Cu(CF3)3 (ref. 5) and (phen)Cu(CF3)3,6 are the most explored copper(III) trifluoromethyl transfer reagents, even though they exhibit limited atom economy in trifluoromethylation.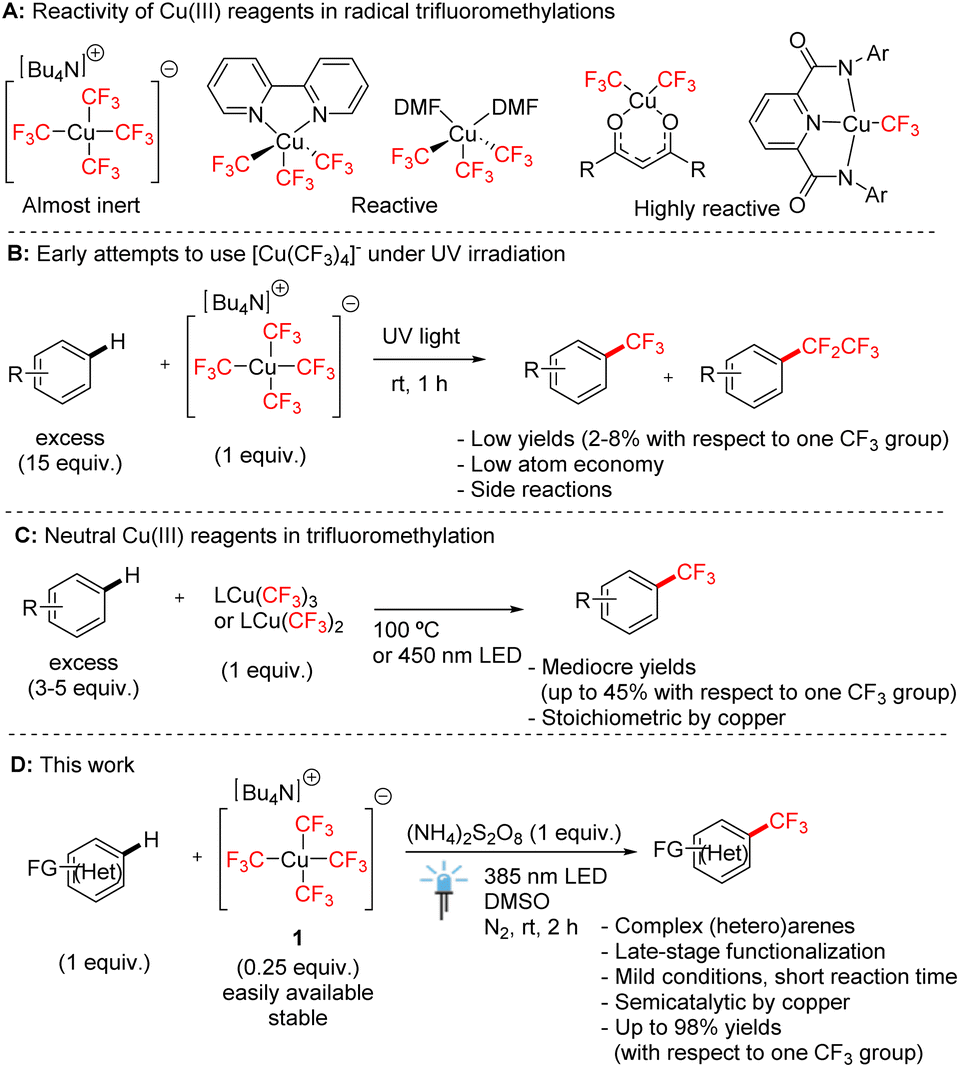 | ||
| Scheme 1 Reactivity of Cu(III) complexes in aromatic C–H trifluoromethylation. | ||
We have recently described oxygen-donor solvate complexes (DMF)2Cu(CF3)3,7 and bis(trifluoromethyl)-1,3-diketonates8 bearing two trifluoromethyl groups which showed high reactivities in trifluoromethylation reactions. Moreover, a highly reactive Cu(III) complex with only one trifluoromethyl group stabilized by a pyridine-2,6-dicarboxamide ligand has recently been reported.9 All these complexes are used as versatile stoichiometric C–H trifluoromethylation reagents under mild conditions (Scheme 1C), the reactivity of which increases with the decreasing number of CF3 ligands. However, no reported transformation starting from Cu(III) trifluoromethyl complexes to date achieved the complete utilization of all trifluoromethyl groups in reactions, highlighting the need for an improved atom efficiency. For example, the yields of trifluoromethylation based on one trifluoromethyl group do not exceed 45% even with the most reactive complexes7 and it is extremely difficult to make use of all CF3 ligands in the complex. We aimed to develop a new strategy that overcomes this limitation. Given that the Bu4N[Cu(CF3)4] salt is the most stable and inexpensive Cu(III) trifluoromethyl species,2,7 a way to utilize it with efficiency would be highly desirable.
Recently, we reported the cleavage of this homoleptic anion by Brønsted acids such as triflic acid, resulting in the formation of solvated Cu(CF3)3 species.7 However, this homoleptic anion is still commonly deemed poorly reactive in radical cleavage and trifluoromethylation.10 Herein, we report a new method of mild light-mediated trifluoromethylation of arenes and heteroarenes, including biomolecules with Bu4N[Cu(CF3)4] in substoichiometric amount (0.25 equiv. of copper) to make use of all four trifluoromethyl groups (Scheme 1D).
Results and discussion
We commenced our studies with attempts to trifluoromethylate 1,3,5-trimethoxybenzene (2), a typical electron-rich arene, with cuprate salt 1 in the most atom-efficient manner possible, employing light as a cost-efficient energy source (Table 1). We were intrigued by the phenomenon of photoinduced homolytic cleavage of Cu(III) trifluoromethyl complexes by blue monochromatic LED light of wavelengths far above their absorption maxima, which is attributed to spin-forbidden HOMO to LUMO + 1 excitation.5c,7 Therefore, we attempted to use an LED source of violet 385 nm light to excite the tetrakis(trifluoromethyl)-cuprate anion in the presence of a persulfate oxidant to facilitate the formation of CF3 radicals. To make use of all four trifluoromethyl groups, we employed a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of arene/[Cu], which would indicate an improvement on the commonly employed stoichiometric Cu–CF3 reagent by making the reaction substoichiometric.
:1 ratio of arene/[Cu], which would indicate an improvement on the commonly employed stoichiometric Cu–CF3 reagent by making the reaction substoichiometric.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%)b |
| a Reaction conditions: 2 (0.4 mmol), 1 (0.25 equiv.), solvent (1 ml), rt, 385 nm LED light, 2 h. b Yields were determined by 19F NMR with PhCF3 as an internal standard and are based on one CF3 group of 1; values in parentheses indicate yields with respect to copper. | ||
| 1 | None | 88 (352) |
| 2 | MeCN as the solvent | 49 (294) |
| 3 | Under air | 81 (325) |
| 4 | K2S2O8 as the oxidant | 84 (337) |
| 5 | Na2S2O8 as the oxidant | 81 (324) |
| 6 | 0.125 equiv. of [Cu] | 50 (400) |
| 7 | No light, 275 nm or 465 nm LED light | <5 |
| 8 | No oxidant | <10 |
| 9 | With Ph4P[Cu(CF3)4] salt | 75 (298) |

After extensive optimization of solvents, oxidants and the reagent ratio (see the SI for full details), we achieved the trifluoromethylation of 2 in 88% yield with respect to the arene and one trifluoromethyl group, which equals to a yield of 352% with respect to the Cu(III) complex (Table 1, entry 1). Switching from dimethylsulfoxide to acetonitrile as the solvent decreased the yield (entry 2), and potassium or sodium persulfates turned out to be slightly less efficient oxidants than ammonium persulfate (entries 4 and 5). The model reaction was only slightly affected by the presence of air (entry 3). Importantly, when the cuprate salt was used in 0.125 equiv. (8:1 arene/[Cu] ratio), the CF3 groups reacted with the substrate quantitatively (400% yield based with respect to copper, entry 6). Control experiments demonstrated that the trifluoromethylation was suppressed in the absence of light or if different irradiation wavelengths were employed, as well as in the absence of an oxidant (entries 7 and 8). The use of tetraphenyl-phosphonium cuprate salt resulted in a slightly lower yield compared to the initially used tetrabutylammonium salt 1 (entry 9).
Under the optimized conditions (Table 1, entry 1), we investigated the method's potential scope of applicability on simple arenes (Scheme 2). High product yields were obtained for electron-rich symmetrical tri- and dimethoxybenzenes (products 3a and 3b). Unsubstituted benzene afforded a moderate yield of product 3c, which matched the reactivity tendency of the electrophilic CF3 radical. Naphthalene predominantly afforded compound 3d produced by β-trifluoromethylation. The method tolerated the presence of the formyl group (product 3e) or the acetamide moiety (3f). Monosubstituted electron-rich arenes mainly afforded mixtures of regioisomers in moderate to good yields (3f–3h), whereas chlorinated biphenyl derivative 3i formed from 1,3,5-trichlorobenzene by radical addition, dimerization, and HCl elimination. The highly electron-deficient substrate dinitrobenzene underwent trifluoromethylation with rather low efficiency (product 3j). Finally, nitrogen heterocycles such as N-phenylpyrrole, 3-methylindole, 2-pyridone and 6-chloropyridazin-3(2H)-one were tolerated giving α-trifluoromethylation products 3k–3n in good yields. Ibuprofen can be also trifluoromethylated in comparatively lower yield. It is worth mentioning that mesitylene, under standard conditions, predominantly afforded the product of radical addition and dimerization (3p), the structure of which has been confirmed by X-ray crystallography. This outcome indirectly supports a radical pathway. Analogous dimerization has been reported previously, although in a different context.13
 | ||
| Scheme 2 Scope of trifluoromethylation of arenes and heteroarenes 2 with tetrakis(trifluoromethyl)cuprate(III) salt 1. aIsolated yields after column chromatography, unless stated otherwise. b 19F NMR yield. | ||
Encouraged by the excellent atom economy and good functional group tolerance of the method, we tested the applicability of copper-mediated trifluoromethylation to the late-stage functionalization of more complex heterocycles (Scheme 3). The anti-gout medication allopurinol was trifluoromethylated smoothly under standard conditions in good yield (product 4). The procedure also proved applicable to xanthine derivatives, such as caffeine and N-propylated theobromine derivative (products 5 and 6). Tryptophan ester bearing amide and indole moieties afforded product 7 also in a good yield. Next, trifluoromethylation of nucleosides and nucleobases was explored. In some cases, adding water as a co-solvent improved the solubility of substrates with unprotected hydroxy groups, demonstrating the water-friendly nature of the present method. Inosine triacetate underwent site-selective trifluoromethylation of the fused imidazole ring (product 8). Uracil and its derivatives smoothly underwent trifluoromethylation to give products 9–11. Azauracil, which is an important inhibitor of DNA synthesis, afforded product 12 in excellent yield. Notable results were obtained for uridine nucleosides, including the synthesis of the well-established anticancer and antiviral drug trifluridine (15) from deoxyuridine as the starting substrate. Finally, brucine was trifluoromethylated in a stereoselective fashion.
 | ||
| Scheme 3 Late-stage trifluoromethylation of complex heterocycles with 1 (0.25 equiv.). aIsolated yields after column chromatography. bDMSO/H2O (3:1) as the solvent and Na2S2O8 as the oxidant. | ||
Having gathered data on a range of trifluoromethylated arenes and heteroarenes, including some prominent biomolecules, we gained further insight into the mechanism of the trifluoromethylation reaction presented. To confirm the key role of the trifluoromethyl radical in this transformation, we performed electron paramagnetic resonance (EPR) analysis of 1 in the presence of spin trap 17 (Fig. 1A). Trifluoromethyl radical adduct 18 was detected by EPR under light irradiation and only an extremely low EPR signal intensity was observed in the absence of light. The crucial role of the light was additionally supported by an on/off experiment (Fig. 1B), in which the reaction was completely shut down upon turning off the irradiation.
 | ||
| Fig. 1 Mechanistic studies of trifluoromethylation with 1. A: EPR detection of the trifluoromethyl radical. B: Lights on/off experiment with a model reaction of trifluoromethylation of 1,3,5-trimethoxybenzene with 1. C: Cyclic voltammetry of 1 (1 mM) in 0.1 M Bu4NPF6 in MeCN at different scan rates (glassy carbon working electrode). D: Spectroelectrochemical investigation of the reduction of 1. E: Absorption spectra upon irradiation of 1. F: Concentration-dependent absorption spectra of 1. G: Absorption spectra during trifluoromethylation of 1,3,5-trimethoxybenzene with 1 according to standard conditions. H: EPR detection of a square planar Cu(II) complex. | ||
The reduction potential of 1 was determined by cyclic voltammetry (Epcvs. SCE = −1.42 V, Fig. 1C). Spectroelectro-chemical analysis of the reduction of 1 showed a decrease in absorbance at 272 nm and the formation of a new absorption band (λmax = 313 nm), with a clear isosbestic point (Fig. 1D). A similar phenomenon was observed by absorption spectroscopy, where the absorption at 313 nm increased after irradiation of 1 at its λmax, albeit with a lower conversion (Fig. 1E). Although absorption of compound 1 shows no overlap with the LED light source used in the trifluoromethylation process (385 nm) at low concentrations, at higher concentrations it exhibits significant absorbance, enabling an effective photoreaction (Fig. 1F). The preparative trifluoromethylation reactions were typically performed at a 0.1 M concentration of 1. Real-time analysis of the absorption spectra during the trifluoromethylation of 1,3,5-trimethoxybenzene with 1 revealed the gradual formation of a new broad band at 600–900 nm (Fig. 1G). This corresponds most likely to the Cu(II)-CF3 species11 and Cu(II) by-products, given that all CF3 groups capable of stabilizing Cu(III) and Cu(I) are consumed in the reaction. EPR analysis following the reaction of 1 in MeCN with the oxidant in the absence of an arene substrate indicated the formation of a Cu(II) complex (most likely symmetrical square planar), and 19F NMR analysis showed the formation of fluoroform, 3,3,3-trifluoropropionitrile, and hexafluoroethane arising from the reaction of trifluoromethyl radicals with hydrogen atoms or fragments of the homolyzed solvent, and dimerization respectively (Fig. 1H).
Based on these observations, we propose that the described trifluoromethylation proceeds by the following mechanism (Scheme 4). Excitation of cuprate salt 1 produces its excited complex 1*, which decomposes into a trifluoromethyl radical and a [CuII(CF3)3]− intermediate. Radical addition to 2 affords radical intermediate A, which upon oxidation to cationic intermediate B and proton transfer to a sulfate anion furnishes product 3. The [CuII(CF3)3]− intermediate can disproportionate to regenerate 1 and form bis(trifluoromethyl)cuprate(I) anion. Oxidation of the Cu(I) species releases the remaining two trifluoromethyl radicals and generates a square-planar Cu(II) sulfate complex 19, corresponding to the complex observed by EPR. The addition of water enabled the isolation of product 20, a known12 light-blue (hence the absorption at 600–900 nm) octahedral hexa–aqua complex 20 that has been characterized by X-ray crystallography (Scheme 4).
 | ||
| Scheme 4 Proposed reaction mechanism of trifluoromethyla-tion of arenes 2 with Bu4N[Cu(CF3)4] (1). | ||
Conclusions
We have developed an efficient and atom-economical method for the C–H trifluoromethylation of (hetero)arenes, including biomolecules, using the readily accessible and stable tetrakis(trifluoromethyl)cuprate(III) salt in substoichiometric amount. This light-driven protocol enables the full utilization of all four CF3 groups, overcoming a long-standing limitation of copper(III) trifluoromethylation chemistry. The reactions proceed under mild conditions and exhibit excellent functional group tolerance, enabling the late-stage functionalization of pharmaceuticals and nucleosides. Mechanistic studies provide evidence that the reactions take place through a radical pathway. These findings expand the synthetic utility of Cu(III) species and set the stage for their future application in organic synthesis.Author contributions
P. P. and V. M. conceived the idea, performed experiments and partially wrote the manuscript, O. M. performed experiments, L. Š. and S. B. performed photochemical and electrochemical measurements, T. S. partially wrote the manuscript, J. T. performed EPR measurements, B. K. performed X-ray analysis, P. B. led the project, obtained funding and partially wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07405c.CCDC 2474866–2474868 contain the supplementary crystallographic data for this paper.14
Acknowledgements
This work was supported by the Czech Academy of Sciences (RVO: 61388963).Notes and references
- (a) X. Liu and Q. Shen, Coord. Chem. Rev., 2021, 442, 213923 CrossRef; (b) H. Valdés and X. Ribas, Organometallic Complexes of Copper, Elsevier, Amsterdam, 4th edn, 2022 Search PubMed.
- (a) A. M. Romine, N. Nebra, A. I. Konovalov, E. Martin, J. Benet-Buchholz and V. V. Grushin, Angew. Chem., Int. Ed., 2015, 54, 2745–2749 CrossRef CAS; (b) D. Naumann, T. Roy, K.-F. Tebbe and W. Crump, Angew. Chem., Int. Ed., 1993, 32, 1482–1483 CrossRef.
- M. Baya, D. Joven-Sancho, P. J. Alonso, J. Orduna and B. Menjón, Angew. Chem., Int. Ed., 2019, 58, 9954–9958 CrossRef CAS.
- (a) B. Zimmer, T. Auth and K. Koszinowski, Chem. – Eur. J., 2023, 29, e202300725 CrossRef CAS PubMed; (b) S. Weske, R. Schoop and K. Koszinowski, Chem. – Eur. J., 2016, 22, 11310–11316 CrossRef CAS PubMed.
- (a) H. R. Zhang, C. C. Feng, N. Chen and S. L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202209029 CrossRef CAS; (b) G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2021, 60, 5467–5474 CrossRef CAS; (c) S. Guo, D. I. AbuSalim and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 11704–11708 CrossRef CAS; (d) S. Guo, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2018, 140, 12378–12382 CrossRef CAS PubMed; (e) W. Paeth, W. Carson, J.-H. Luo, D. Tierney, Z. Cao, M.-J. Cheng and W. Liu, Chem. – Eur. J., 2018, 24, 11559–11563 CrossRef; (f) P. Zhang, H. Chen, L. Zhu, W. Cao and C. Li, Org. Lett., 2018, 20, 7062–7065 CrossRef CAS.
- (a) S.-L. Zhang, H.-X. Wan and W.-F. Bie, Org. Lett., 2017, 19, 6372–6375 CrossRef CAS; (b) S.-L. Zhang and W.-F. Bie, RSC Adv., 2016, 6, 70902–70906 RSC; (c) N. Chen and S.-L. Zhang, Adv. Synth. Catal., 2022, 364, 3941–3947 CrossRef CAS; (d) J.-J. Dong and S.-L. Zhang, Adv. Synth. Catal., 2020, 362, 795–800 CrossRef CAS.
- V. Motornov, M. Procházka, N. Alpuente, P. Salvador, P. Slavíček, B. Klepetářová and P. Beier, ChemistryEurope, 2024, 2, e202400004 CrossRef CAS.
- V. Motornov, B. Klepetářová and P. Beier, Adv. Synth. Catal., 2023, 365, 2858–2864 CrossRef CAS.
- M. A. P. Ball, P. J. Myers, G. D. Ritch, J. K. Bower, C. E. Moore, N. K. Szymczak and S. Zhang, Angew. Chem., Int. Ed., 2024, 64, e202420677 CrossRef PubMed.
- P. Alayoglu, T. Chang, C. Yan, Y.-S. Chen and N. P. Mankad, Angew. Chem., Int. Ed., 2023, 62, e202313744 CrossRef CAS PubMed.
- Y. Weng, Y. Jin, J. Wu, X. Leng, X. Lou, F. Geng, B. Hu, B. Wu and Q. Shen, J. Am. Chem. Soc., 2024, 146, 23555–23565 CrossRef CAS PubMed.
- B. N. Figgis, A. N. Sobolev, C. J. Simmons, M. A. Hitchman, H. Stratemeier and M. J. Riley, Acta Crystallogr., 2000, B56, 438–443 CAS.
- S. Karreman, S. B. Karnbrock, S. Kolle, C. Golz and M. Alcarazo, Org. Lett., 2021, 23, 1991–1995 CrossRef CAS.
- (a) CCDC 2474866: Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p29dh ; (b) CCDC 2474867: Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p29fj ; (c) CCDC 2474868: Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p29gk .
| This journal is © The Royal Society of Chemistry 2026 |