
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation of tungsten ethylidene complexes from diethyl complexes through a proton-catalyzed rearrangement of ethylene
Milan
Maji
,
Landley
Zeng
,
Richard R.
Schrock
 *,
Matthew P.
Conley
* and
Veronica
Carta
*,
Matthew P.
Conley
* and
Veronica
Carta
Department of Chemistry, University of California, Riverside, California 92521, USA. E-mail: richard.schrock@ucr.edu
First published on 12th November 2025
Abstract
W(NAr)2Et2 (Ar = 2,6-diisopropylphenyl) reacts with two equivalents of RF9OH (ORF9 = OC(CF3)3) to yield W(NAr)(ArNH2)(ORF9)2(C2H4) complexes and ethane. In solution W(NAr)(ArNH2)(ORF9)2(C2H4) decomposes to give RF9OH, ethane, W(NAr)(ORF9)2(C2H4), and W(NAr)(NHAr′)(ArNH2)(ORF9), in which Ar′ contains a dehydrogenated isopropyl group (Ar′ = (2-i-Pr)(6-CMe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)C6H3) coordinated to the metal. On a similar time scale W(NAr)(ORF9)2(CHCH3) complexes are formed from W(NAr)(ORF9)2(C2H4) through an ArNH2-catalyzed rearrangement of the ethylene ligand. W(NAr)(NHAr′)(ArNH2)(ORF9) reacts with cyclohexene to form methylenecyclohexene and complexes that contain an NHAr″ ligand where Ar″ is a disubstituted (methyl/aryl) alkylidene, (2-i-Pr)(6-CMe)C6H3) that is tethered to the metal through the amido nitrogen. In contrast to W(NAr)(ArNH2)(ORF9)2(C2H4), analogous ORF6 (OCMe(CF3)2) and ORF3 (OCMe2(CF3)) complexes are relatively stable at 22 °C.
CH2)C6H3) coordinated to the metal. On a similar time scale W(NAr)(ORF9)2(CHCH3) complexes are formed from W(NAr)(ORF9)2(C2H4) through an ArNH2-catalyzed rearrangement of the ethylene ligand. W(NAr)(NHAr′)(ArNH2)(ORF9) reacts with cyclohexene to form methylenecyclohexene and complexes that contain an NHAr″ ligand where Ar″ is a disubstituted (methyl/aryl) alkylidene, (2-i-Pr)(6-CMe)C6H3) that is tethered to the metal through the amido nitrogen. In contrast to W(NAr)(ArNH2)(ORF9)2(C2H4), analogous ORF6 (OCMe(CF3)2) and ORF3 (OCMe2(CF3)) complexes are relatively stable at 22 °C.
Introduction
Several recent papers1–10 have begun to provide answers to long-standing questions in the area of olefin metathesis11 that concern how molybdenum or tungsten alkylidene complexes, MCRR′ (R or R′ = H or an alkyl), are formed from olefins, in solution, or in metal complexes deposited on a support such as silica.12 Two mechanisms have now been documented, mostly through synthetic and mechanistic studies of complexes in solution. One mechanism consists of a photochemical ring-contraction13 of a metallacyclopentane14 formed from two olefins in the presence of blue light (450 nm) to give a metallacyclobutane, a key intermediate in metathesis, or a terminal alkylidene through α hydrogen abstraction15–17 within the MC4 ring.2,6,10 The second mechanism consists of a proton-catalyzed olefin rearrangement (an “Hcat” reaction18,19), i.e., addition of a proton to a bound alkene to give an intermediate alkyl that contains both α and β protons, followed by loss of an α proton to give an alkylidene.9 So far it has been shown that a proton can be provided by an external cationic acid (anilinium1,4,5) or by an amine (RNH2) upon binding to an electron-poor tungsten center.9
Addition of two equivalents of RF9OH or RF6OH to W(NAr)2R2 complexes (R = n-Pr or i-Pr) has been found to lead to double protonation of the imido ligand and either propylene or isopropylidene complexes that interconvert in a ArNH2-catalyzed (Hcat) reaction; no n-propylidene complexes were observed.9 It was proposed that these Hcat reactions are under thermodynamic control and that although terminal alkylidenes could be formed, they are not because isopropylidene complexes are at least 2–3 kcal mol−1 lower in energy than n-propylidene complexes in otherwise identical compounds. Because some of the earliest studies in metathesis concerned the synthesis of olefin metathesis initiators from main group alkylating agents such as ethyl aluminum compounds,20–23 an obvious question is can an ethylidene ligand be formed similarly upon addition of RF9OH to W(NAr)2Et2, and if so, by what mechanism? The results reported here attempt to answer these questions.
Results
Protonation of W(NAr)2Et2
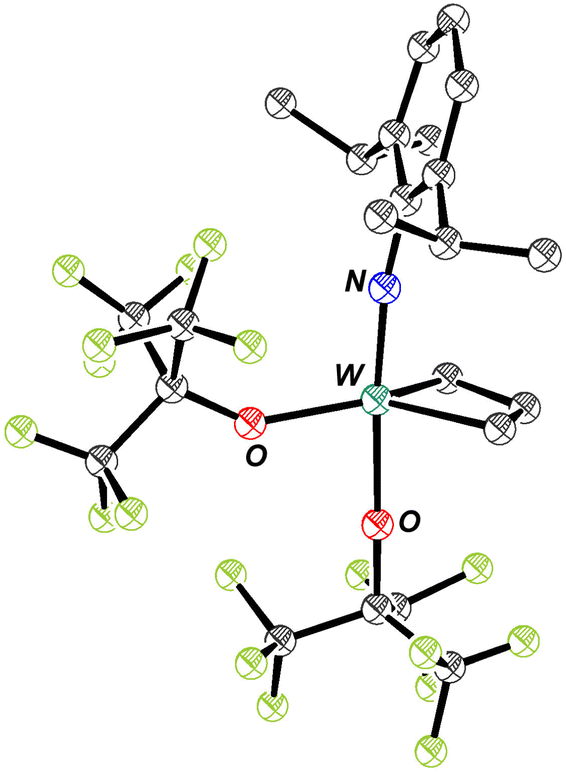
W(NAr)2Et2 can be made in good yield through addition of ethyl Grignard to W(NAr)2Cl2(1,2-dimethoxyethane). It has a metallocene-like24–27 structure (see Fig. 1) in which there is no evidence for either an α or a β agostic CH interaction28,29 in an ethyl group. Only three W–N π interactions are likely in W(NAr)2Et2, leaving one occupied ligand-centered non-bonding orbital (LCNBO) at a relatively low energy that is susceptible to electrophilic attack.30–32 | ||
| Fig. 1 The molecular structure of W(NAr)2Et2 as determined by SCXRD (hydrogen atoms have been omitted for clarity). | ||
Addition of two equivalents of RF9OH to a C6D6 solution of W(NAr)2Et2 yields a complex, but decipherable, mixture of species that evolves over time. The reactions that are supported by experimental data are summarized in eqn (1)–(3). Compound 1 forms first by what appears to be overall a double protonation of an imido ligand in W(NAr)2Et2 and β abstraction to give ethane in a diethyl intermediate. Compound 1 could be isolated through crystallization from pentane in 52% yield, but subsequent crops were contaminated with 2 and 3. An X-ray study shows that the structure of 1 is closest to a square pyramid with ethylene in the apical position and the ORF9 ligands trans to the imido and ArNH2 ligands in basal positions (Fig. 2).
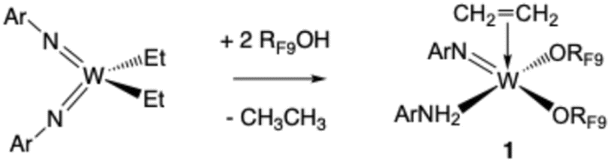 | (1) |
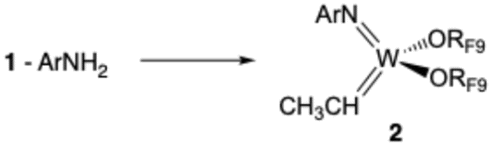 | (2) |
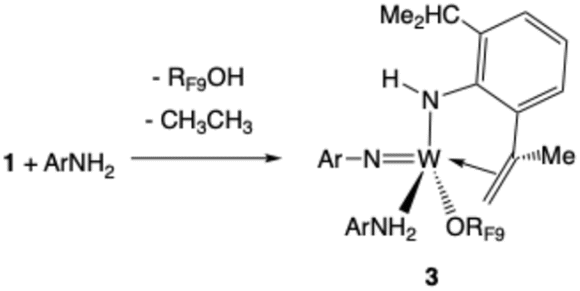 | (3) |
 | ||
| Fig. 2 The molecular structure of W(NAr)(ArNH2)(ORF9)2(C2H4) (1) as determined by SCXRD. | ||
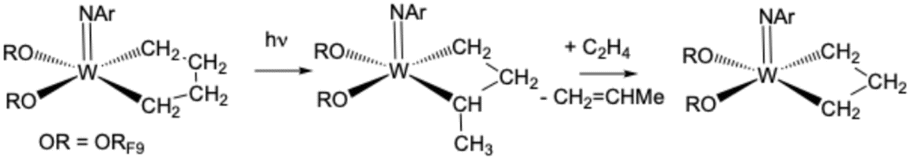
The formation of 2, 3, and a methylene complex (4; W(NAr)(CH2)(ORF9)2) in a typical reaction between RF9OH and W(NAr)2Et2 can be followed in the proton NMR spectral region between 8.6 and 11.8 ppm (Fig. 3). The two quartets for anti and syn Hα protons in isomers of W(NAr)(ORF9)2(CHCH3) (2) begin to appear in minutes at 11.72 ppm and 11.16 ppm, respectively. These isomers form at approximately the same rate, with the syn isomer dominating after 22 h, probably through isomerization of anti to the lower energy syn isomer,33 either through rotation about the WC bond or through a protonation/deprotonation sequence that involves formation of an intermediate ethyl complex. Complex 3, whose NH proton is observed at 8.8 ppm, forms at approximately the same rate as the ethylidene complexes. Finally, two resonances for inequivalent protons in a methylene complex, most likely W(NAr)(ORF9)2(CH2), begin to appear after ∼1 h. The relative amounts of 2, 3, and 4 vary somewhat from run to run, but in a spectrum similar to the top one in Fig. 3 the amounts versus a 1,4-bis-TMS-benzene standard at room temperature after 24 h are 11% 2, 32% 3, and 13% 4 relative to the amount of starting 1.
 | ||
| Fig. 3 Partial 1H NMR spectra showing the conversion of W(NAr)2Et2 to W(NAr)(NHAr′)(ArNH2)(ORF9) (3) (##), syn and anti ethylidene complexes (CHMe*) (2), and a methylidene complex (CH2#) (4). | ||
It is clear that the ArNH2 ligand is labile in 1 and that ArNH2 is required for forming 2 catalytically (eqn (2)) and for forming 3 irreversibly (eqn (3)). NMR studies show that the ArNH2 ligand in 1 is bound on the NMR time scale at low temperatures but dissociates at room temperature to give W(NAr)(ORF9)2(ethylene), an analog of W(NAr)(OSiPh3)2(ethylene).8 Therefore, addition of one equivalent of B(C6F5)3 to 1 in solution gives a mixture of W(NAr)(ORF9)2(C2H4) and (ArNH2)[B(C6F5)3] (Fig. S36). W(NAr)(ORF9)2(C2H4) was isolated as an orange oil in 61% yield after careful removal of crystalline (ArNH2)[B(C6F5)3] from −30 °C pentane solutions, but crystals of W(NAr)(ORF9)2(C2H4) could not be obtained. A key fact is that W(NAr)(ORF9)2(C2H4) is relatively stable in solution in the absence of ArNH2 (see S36–S38). However, when ArNH2 is reintroduced into a solution of pure W(NAr)(ORF9)2(C2H4), mixtures that contain 2, 3, and 4 again form that are similar to those shown in Fig. 3. ArNH2 is required for rearrangement of 1 to 2, but it is consumed to form 3. As with the recently reported interconversion of propylene and isopropylene complexes catalyzed by ArNH2,9 details of these reactions are not clear, but it seems likely that ArNH2 coordinates to tungsten before a proton transfers to another ligand in an Hcat reaction. The fact that the pKa of anilinium and RF9OH are similar (∼5) raises the possibility that anilinium could be a proton shuttle in some situations.
We propose that the ethylidene complexes form through an ArNH2-catalyzed rearrangement of the ethylene to an ethylidene ligand; an ethyl complex is the proposed intermediate. As noted earlier, W(NAr)(ORF9)2(C2H4) is relatively stable at room temperature in solution. We assume that the ethylidene complexes do not contain coordinated ArNH2, as ArNH2 is consumed to make W(NAr)(NHAr′)(ArNH2)(ORF9). We did not attempt to isolate the ethylidene complexes because 14e tungsten complexes of this general type are prone to decompose bimolecularly to give W2 dimers when the terminal alkylidene is relatively small.34,35 We could find no example of an isolated 14e tungsten ethylidene complex of the type discussed here in the literature, although the stability of an ethylidene toward bimolecular decomposition will depend, inter alia, upon the degree of steric protection against bimolecular decomposition that is provided by large imido and alkoxide ligands (see also the Discussion section).
Compound 3 is formed when ArNH2 in solution attacks 1. One Ar isopropyl group is dehydrogenated to give the Ar′ group and ethane. Compound 3 could be isolated as red crystals. An X-ray study (Fig. 4) showed that the C(Me)CH2 group in the NHAr′ ligand so formed is bound to the metal. In a typical NMR spectrum the yield of W(NAr)(NHAr′)(ArNH2)(ORF9) relative to a 1,4-bis TMS benzene internal standard was of the order of 30% (Fig. S34). We propose that CH activation in the isopropyl group takes place in a W(NHAr) complex and is possible because the NHAr ligand is bent, which brings an isopropyl group closer to the metal than in a NAr ligand and thus facilitates CH activations in it. Intimate details of this reaction are unclear.
 | ||
| Fig. 4 The molecular structure of W(NAr)(NHAr′)(ArNH2)(ORF9) as determined by SCXRD. | ||
The results presented so far suggest that the proton in coordinated ArNH2 is relatively acidic. Therefore it could be removed by an external base. 2,6-Lutidine (Lut; two equivalents) reacts with 1 to form W(NAr)2(ORF9)(CH2CH3) and (LutH)(ORF9) (eqn (4)), according to NMR data (Fig. S68 and 69).
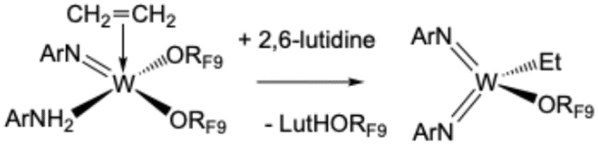 | (4) |
The proton resonance in (LutH)(ORF9) is found at 14 ppm in the reaction mixture. No ethylidene complex was observed. Although we again do not know the precise sequence of reactions that leads to the result shown in eqn (4), it is clear that the protons in a bound ArNH2 are removed, one is transferred to lutidine to yield lutidinium as a perfluoro-t-butoxide salt and the other is transferred to the ethylene ligand. Movement of a proton from N to C is related to protonation of bound styrene by PhNMe2H+1,5 and to intermediates formed in the reaction of W(NAr)2Et2 with RF9OH. For reference it should be noted that the pKa of LutH+ is ∼9 in water, while that for ArNH3+ is ∼5.
It should be noted that addition of two equivalents of RF6OH or RF3OH to W(NAr)2Et2 leads to formation of only W(NAr)(ArNH2)(ORF6)2(ethylene) (54% isolated) and W(NAr)(ArNH2)(ORF3)2(ethylene) (76% isolated as an oil), respectively. Both are stable in solution at 22 °C (Fig. S52) toward the type of reactions described for RF9OH.
Reactions of 1 with ethylene
W(NAr)(ArNH2)(ORF9)2(ethylene) reacts immediately with ethylene to give the 14e square pyramidal metallacyclopentane complex, W(NAr)(ORF9)2(C4H8) (eqn (5) and Fig. 5), the only tungsten complex observed, and free ArNH2 (Fig. S42 and S49). W(NAr)(ORF9)2(C4H8) is also obtained as the only product upon treatment of W(NAr)(ORF9)2(ethylene) with ethylene (Fig. S46). A square pyramidal structure for the tungstacyclopentane in which the imido ligand is in an apical position is the only five-coordinate structural type that has been observed. Upon irradiation of W(NAr)(ORF9)2(C4H8) with 450 nm light under ethylene the trigonal bipyramidal isomer of W(NAr)(ORF9)2(C3H6) (Fig. 6 and S51) and propylene are formed (eqn (6)), as has been found for W(NAr)(OSiPh3)2(C4H8).2 Unsubstituted tungstacyclobutane complexes with a TBP structure analogous to that found for W(NAr)(ORF9)2(C3H6) have been observed when relatively electron-withdrawing alkoxides are present. SP tungstacyclobutane complexes, including substituted versions, have been observed when the alkoxides are more electron-donating (OCMe3 or OC(CF3)Me2).17,36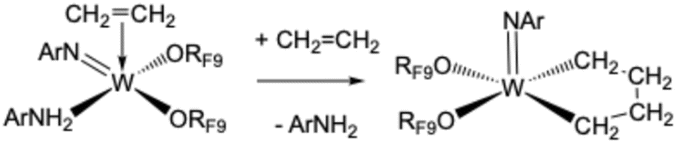 | (5) |
 | (6) |
 | ||
| Fig. 5 The molecular structure of W(NAr)(ORF9)2(C4H8) as determined by SCXRD. | ||
 | ||
| Fig. 6 The molecular structure of W(NAr)(C3H6)(ORF9)2 as determined by SCXRD. | ||
Because tungsten imido ethylene complexes are not common, it would be desirable to make one that can be used to make others in the absence of aniline. In a reaction analogous to that reported for Mo(NAr)(OTf)2(C2H4)(dme),37 W(NAr)(CHCMe2Ph)(OTf)2(dme) was treated with ethylene (60 psi) in toluene for two days at 80 °C. W(NAr)(OTf)2(C2H4)(dme) was isolated as an orange powder in ∼50% yield upon filtration of the reaction mixture (Fig. S83–86). A variety of aniline-free ethylene derivatives should be preparable from W(NAr)(OTf)2(C2H4)(dme).
Reactions of 3 with ethylene and cyclohexene
W(NAr)(NHAr′)(ArNH2)(ORF9) reacts immediately upon diffusion of ethylene into a solution containing it to yield a deep red compound that we propose to be the tricyclic complex shown in eqn (7), and free ArNH2 (Fig. S72–S76). This tricyclic compound is unchanged when irradiated with 450 nm LED light in the absence or presence of ethylene, unlike other unsubstituted or substituted tungstacyclopentanes of a similar type.2,4,6 It is stable in benzene in the presence of ethylene, but it reverts to W(NAr)(NHAr′)(ArNH2)(ORF9) when ethylene is removed from the sample in vacuo (Fig. S77). It has not been isolated so far for that reason. The 13C chemical shifts for the α carbon atoms in the WC4 ring in W(NAr)(Ar′NHC2H4)(ORF9) are 95.1 ppm (for CαMe) and 72.9 ppm (for CαH2; Fig. S76). The Cβ resonances are found at 50.78 ppm (for Cβ next to CαMe) and 36.84 ppm (for Cβ next to CH2).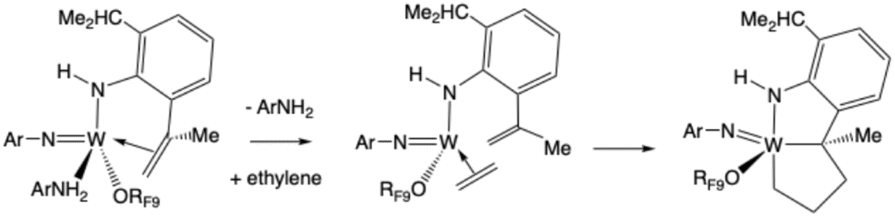 | (7) |
W(NAr)(NHAr′)(ArNH2)(ORF9) also reacts with cyclohexene at 100 °C to give methylene cyclohexane and a deep purple solution that contains a complex in which there is an alkylidene ligand with a Cα resonance at 214.8 ppm (Fig. S82). All NMR data are consistent with the purple complex being the monomer shown in Scheme 1 or a dimeric version of it (see discussion immediately below). We propose that cyclohexene replaces ArNH2 and the bound olefin in the NHAr′ ligand and is converted into a cyclohexylidene ligand in an ArNH2-catalyzed reaction.9 A metathesis reaction between the cyclohexylidene and the CMeCH2 group then yields methylenecyclohexane (<50% according to NMR studies) (Fig. S79) and a compound that contains a tethered alkylidene (the NHAr″ ligand; Scheme 1).
 | ||
| Scheme 1 The proposed mechanism of formation of methylenecyclohexene and W(NAr)(NHAr″)(ORF9)(ArNH2). | ||
All efforts to crystallize a product from the reaction mixture shown in Scheme 1 failed. Therefore, four equivalents of pyridine were added to the purple crude reaction mixture in an effort to replace ArNH2 in the proposed complex shown in Scheme 1 with a more strongly bound pyridine ligand. The mixture turned deep red and crystals began to form after 1 h. A dimeric complex (Fig. 7) is formed through the proposed reaction shown in eqn (8) in which the NHAr″ ligand (lower right) is now bridging two tungsten centers with W–N distances of 2.14 and 2.32 Å and the NHAr′ ligand has lost its NH proton to yield RF9OH and an unsymmetrically bridging NAr′ ligand (upper left); the W–N distances are 1.83 and 2.32 Å.
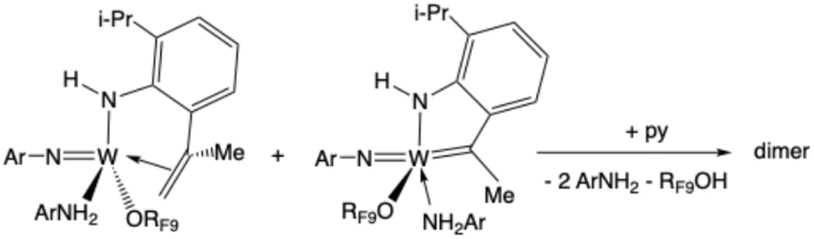 | (8) |
 | ||
| Fig. 7 The molecular structure of W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9) as determined by SCXRD. | ||
The W–C bond lengths in the bridging NHAr″ ligand shown in Fig. 7 are 2.139 Å and 2.294 Å, which are similar to the W–C bond lengths in the µ-CH2 ligands found in [W(NArCl)(Biphen)(µ-CH2)]2 (ArCl = 2,6-dichlorophenyl).38 The W–C distances are 2.27 Å and 1.98 Å for a difference of 0.29 Å compared to a difference of 0.16 Å in the compound in Fig. 7. In [W(NAr)(Biphen)(µ-CH2)]2 the Cα resonance is found at 186 ppm with JCW values of 79 and 37 Hz.39 The alkylidene Cα resonance in the dimer shown in Fig. 7 was found at 301.4 ppm (Fig. S26 and S82). We propose that the initial purple product formed from W(NAr)(NHAr′)(ArNH2)(ORF9) is a dimer related to that shown in Fig. 7, perhaps one in which a labile ArNH2 ligand is bound in place of the pyridine in the isolated complex.
A chemical shift of 301.4 ppm for the alkylidene carbon atom in W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9) is more indicative of a nonbridging alkylidene. Therefore, DOSY NMR studies on W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9) were carried out in benzene-d6 (see SI). Those studies showed that a single compound is present (Fig. S31) that has a molecular weight within experimental error of that corresponding to the dimer shown in Fig. 7. On this basis the simplest explanation is that a chemical shift of 301.4 ppm for the bridging alkylidene carbon atom in W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9) is correct. However, we favor the possibility that W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9) has isomerized to yield a dimer in which the alkylidene Cα is no longer bridging two metal centers. The main point, however, is that the NAr″ framework that contains a disubstituted (CHMe) tethered alkylidene has been formed with the mechanism being a metathesis reaction that yields methylenecyclohexane, as shown in Scheme 1.
Discussion and conclusions
The reactions reported here are part of an exploration of proton transfers between C, N, and O atoms in the first coordination sphere of high oxidation state organometallic Mo and W complexes relevant to olefin metathesis that began with the isomerization of styrene complexes to phenethylidene complexes.1,4,5 The present study was aimed at determining whether ethylidene complexes could be formed through proton migrations between C, N, and O atoms in a manner similar to reactions where internal alkylidenes are formed from propyl9 or cyclohexyl7,8 ligands. We have found here that ethylidene complexes indeed can be formed from diethyl complexes, but ethylene complexes are formed first followed by an ArNH2-catalyzed isomerization of ethylene to ethylidene complexes. No ethylidene complexes are observed at 22 °C when the added alcohol is RF6OH or RF3OH, presumably because protons from a coordinated aniline are much more mobile in perfluoro-t-butoxide complexes.A 10% yield of ethylidenes from diethyl complexes would be more than enough to account for the catalytic metathesis activities observed in “classical” metathesis systems in which the alkylidene initiators are formed upon addition of a main group ethylating reagent. Formation of ethylidenes through an α abstraction or deprotonation reaction of course cannot be excluded in other circumstances, but in the chemistry reported here an ArNH2-catalyzed rearrangement of an ethylene ligand seems far more likely to be the way ethylidene complexes are formed.
The possible roles that bimolecular decomposition reactions in solution to give metal–metal bonded complexes can play in high oxidation state organometallic tungsten chemistry have not been fully explored in the literature, in part because of the almost endless number of possibilities. The ethylene required for metathesis reactions that we proposed earlier as the origin of methylene complexes could be formed through bimolecular decomposition of ethylene complexes to give W2 dimers. An example is the following (Ar′ is 2,6-dimethylphenyl in this case):40 “Upon heating {W(NAr′)[OCMe2(CF3)]2}2(C2H4) in toluene to 80 °C, it loses ethylene to yield {W(NAr′)[OCMe2(CF3)]2}2, but also is transformed into a new species that has C1-symmetry and that contains an ethyl group as a consequence of activation of an ortho methyl group in the NAr′ ligand” (eqn (9) is taken directly from ref. 40). The complex with C1-symmetry is the W(V) product shown in eqn (9) that contains two bridging imido groups. Both dimers shown in eqn (9) were crystallographically characterized.
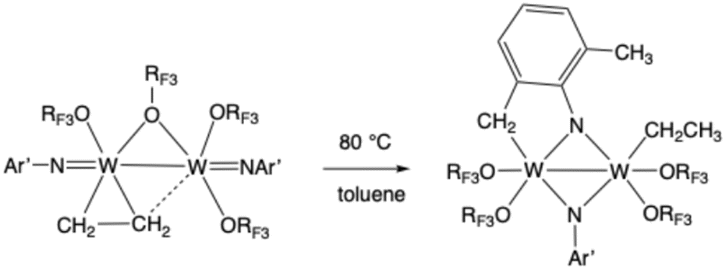 | (9) |
The chemistry described in the quote and in eqn (9) is similar to the chemistry reported in this paper in that it includes proton transfers between C, N, and O atoms in the first coordination sphere (see also the synthesis and X-ray structure of [W(NArCl)(Biphen)]2(µ-CH2CH2), in which an ethylene carbon is bound to each tungsten39). It also should be noted that tungsten–tungsten complexes can initiate olefin metathesis reactions.40 Although bimolecular chemistry may be slow relative to monometallic chemistry, it must be considered to be a source of products over the long term in metathesis chemistry in solution, even to the extent of reforming free ethylene through a complex decomposition of ethylidenes that may involve chemistry of tungsten–tungsten dimers.
Bridging, high oxidation state alkylidenes of Mo and W are rare. We are aware of two that have been reported.38,39 Both are bridging methylenes; one of the two has been crystallographically characterized. The bridging alkylidene complex shown in Fig. 7 is a third example. Because a substituted alkylidene cannot form an alkylidyne ligand, it could eventually be an initiator in a metathesis reaction upon rearrangement of a dimer to an isomer that contains a nonbridging alkylidene or to yield a monomeric alkylidene complex. The disubstituted bridging alkylidene found in the structure shown in Fig. 7 is the only one reported to our knowledge. The possibility that the dimer in Fig. 7 may isomerize to one that contains a nonbridging alkylidene (with Cα at 301.4 ppm) suggests that reactive disubstituted alkylidene complexes may be available from dimers.
The diisopropylphenylimido (NAr) ligand has been a mainstay of much metathesis-related imido chemistry of Mo and W, largely because it has been stable toward unimolecular or bimolecular side reactions that involve it. But as we have shown here, an o-isopropyl group in the Ar group can be dehydrogenated. The dehydrogenation probably takes place in bound ArNH2 and/or ArNH ligands, because in both, the M–N–Cipso angle is much smaller than in NAr ligands (where it is typically >160°). A decomposition of Mo(NAr)(CH2CH2)(OAr)2 to give Mo(NAr)(OAr)(OAr′)(Et2O) is related to the results described here.5
The sequence of reactions shown in Scheme 1 are proposed to lead to the unusual tethered internal alkylidene as a consequence of formation of a cyclohexylidene ligand from cyclohexene. High oxidation state tungsten alkylidenes that are tethered to a covalently bound ligand are gaining increased attention in metathesis chemistry because of the mechanistic restrictions that such designs can impose.41–47 Although the method of forming the tethered NHAr″ alkylidenes described here is relatively exotic, simpler, more accessible methods could be developed for making them and variations of them.
Experimental section
General considerations
Unless otherwise stated, all manipulations were carried out using standard Schlenk or glovebox techniques under an N2 atmosphere. Pentane, diethyl ether, dichloromethane, benzene and toluene were dried and deoxygenated by argon purge followed by passage through activated alumina in a solvent purification system and storage over 4 Å molecular sieves. Solvents that do not contain halides or CN groups were tested with a purple solution of sodium benzophenone ketyl in THF to confirm effective oxygen and moisture removal prior to use. W(NAr)2Cl2(dme) was prepared according to a reported procedure.1 Perfluoro-t-butanol and hexafluoro-t-butanol were stored over activated 4 Å molecular sieves. 1,4-TMS benzene, isopropyl magnesium chloride, and n-propyl magnesium chloride were purchased from Sigma-Aldrich. B(C6F5)3 was purchased from Alfa chemical and sublimed before use. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Deuterated solvents were purchased from Cambridge Isotope Laboratories Inc. They were degassed and dried over activated 4 Å molecular sieves for at least 24 h prior to use. NMR spectra were recorded on Bruker Avance 600 MHz and Bruker Avance 500 MHz spectrometers. 1H and 13C chemical shifts are reported in ppm relative to tetramethylsilane using residual solvent as an internal standard. 19F chemical shifts are reported in ppm relative to trichlorofluoromethane as an external standard. NMR data can be found in the SI.Synthesis of W(NAr)2Et2
W(NAr)2Cl2(dme) (1.00 g, 1.50 mmol), a magnetic stir bar, and diethyl ether (20 mL) were added to a 100 mL round bottom flask. The flask was cooled to −116 °C and a −116 °C solution of ethyl magnesium bromide (0.40 g, 3.01 mmol, 2 equiv.) in 5 mL of diethyl ether was added over a period of 3 min in a dropwise manner. The orange solution immediately turned yellow, and a white precipitate began to form. The mixture was warmed to room temperature over 3 h. All volatiles were then removed in vacuo and the residue was then extracted in pentane (3 × 5 mL) and the mixture was filtered through Celite. Pentane was removed in vacuo and the solid residue was redissolved in minimal pentane (∼4 mL). Two batches of orange crystals were obtained upon storing the pentane solution in a freezer at −30 °C overnight; yield 0.773 g, 87%. A reaction on a scale of 4–5 g led to a decreased yield of ∼50%. Anal. calcd for C28H44N2W: C, 56.76; H, 7.49; N, 4.73. Found C, 56.32; H, 7.59; N, 4.77 (see SI for complete NMR data).Synthesis of W(NAr)(ORF9)2(C2H4)(ArNH2)
W(NAr)2Et2 (1.60 g, 2.70 mmol), a magnetic stir bar, and pentane (20 mL) were added to a 100 mL round bottom flask. The flask was cooled to −130 °C and a solution of RF9OH (1.34 g, 5.67 mmol, 2.10 equiv.) in 5 mL of pentane was added dropwise over a period of 3 min. The mixture was allowed to warm to room temperature over a period of 30 min. The color changed from orange-yellow to red. All volatiles were then removed in vacuo and the solid residue was dissolved in ∼3 mL of pentane. One batch of orange-red crystalline solid was obtained upon storing the pentane solution at −30 °C overnight; yield 1.47 g, 52%. Only the first crop of crystals should be collected within one day, as subsequent crops contain increasing amounts of W(NAr)(Ar′NH)(ArNH2)(ORF9) (see below). Anal. calcd for WC34H40F18N2O2W: C, 39.47; H, 3.90; N, 2.71. Found C, 40.12; H, 4.18; N, 2.46.Synthesis of W(NAr)(ORF9)2(C2H4)
In a 20 mL glass vial, W(NAr)(ORF9)2(C2H4)(ArNH2) (300 mg, 0.299 mmol), B(C6F5)3 (148 mg, 0.299 mmol, 1 equiv.), and pentane (5 mL) were added. This solution was stirred at room temperature for 1 h. The volume was reduced to ∼3 mL and kept at −30 °C overnight. ArNH2(B(C6F5)3) precipitated as a white crystalline solid and the mother liquor was quickly pipetted out. This process was repeated twice. The final product was obtained as an orange oil; yield 152 mg (61%). No crystals have been obtained.Synthesis of W(NAr)(ORF9)2(C4H8)
W(NAr)(ORF9)2(C2H4)(ArNH2) (80 mg, 0.077 mmol) was dissolved in benzene (3 mL) in a 25 mL storage flask and the flask was sealed and subjected to three freeze-pump-thaw cycles. This solution was exposed to an ethylene atmosphere (15 psi) at room temperature and stirred with a magnetic stir-bar. The color changed immediately from orange to yellow. All volatiles were removed in vacuo and the yellow orange oil was dissolved in pentane (∼1.0 mL) and the solution transferred to a 20 mL glass vial and placed inside the freezer at −30 °C of glovebox. X-ray quality yellow crystals of the desired compound were obtained after 1 day from pentane. NMR spectra showed W(NAr)(ORF9)2(C4H8) to be the only tungsten product. The same result was obtained in a reaction between W(NAr)(ORF9)2(C2H4) and ethylene.Synthesis of W(NAr)(ORF9)2(C3H6)
W(NAr)(ArNH2)(ORF9)2(C2H4) (50 mg, 0.048 mmol) was dissolved in a mixture of pentane (0.5 mL) and toluene (0.1 mL) and the solution was transferred to a J. Young NMR tube that was sealed and subjected to freeze–pump–thaw three times before adding ethylene (15 psi). The color of the solution of the solution changed from orange to yellow, as described above. This tube was then irradiated with blue LED (Kessil LED at 450 nm) for 2 h at ambient temperature. NMR examination of a portion of the product from which all solvent had been removed showed that only W(NAr)(ORF9)2(C3H6) and ArNH2 were present. The tube was kept inside the freezer at −30 °C to give X-ray quality yellow crystals of W(NAr)(ORF9)2(C3H6) after 3 days.Synthesis of W(NAr)(ORF6)2(C2H4)(ArNH2)
This synthesis is similar to that for W(NAr)(ORF9)2(C2H4)(ArNH2) from W(NAr)2Et2 (584 mg, 0.980 mmol) and a solution of RF6OH (233 mg, 2.07 mmol, 2.1 equiv.) in 10 mL of pentane. The solid residue was recrystallized from minimal pentane (∼3 mL). One batch of orange crystalline solid was collected after storing the pentane solution at −30 °C overnight; yield 494 mg (54%). Anal. calcd for WC34H46F12N2O2W: C, 44.07; H, 5.00, 3.02. Found C, 43.68; H, 5.10; N, 2.99.Synthesis of W(NAr)(ORF3)2(C2H4)(ArNH2)
This synthesis is similar to that for W(NAr)(ORF9)2(C2H4)(ArNH2) from W(NAr)2Et2 (600 mg, 1.01 mmol) and a solution of RF3OH (273 mg, 2.12 mmol, 2.10 equiv.) in 10 mL of pentane. After 3.5 h all volatiles were removed in vacuo to give the final product as an orange liquid; yield 630 mg (76%).Synthesis of W(NAr)(Ar′NH)(ArNH2)(ORF9)
After collecting crystals of W(NAr)(ORF9)2(C2H4)(ArNH2) in the synthesis of W(NAr)(ORF9)2(C2H4)(ArNH2) described above, the pentane solution was concentrated and kept at −30°. After 24 h two crops of red crystalline W(NAr)(Ar′NH)(ArNH2)(ORF9) were collected; yield 410 mg (16%). Anal. calcd for WC40H52F9N3OW: C, 50.80; H, 5.54; N, 4.44. Found C, 50.91; H, 5.56; N, 4.39.Synthesis of W(NAr)2(ORF9)(CH2CH3)
W(NAr)(ORF9)2(C2H4)(ArNH2) (200 mg, 0.190 mmol) was treated with 2,6-lutidine (45 mL, 0.38 mmol) in benzene (6 mL) in a 20 mL glass vial for 16 h. All volatiles were removed in vacuo and the crude residue was extracted into pentane (∼4 mL). This solution was kept under vacuum for 3 h to remove excess 2,6-lutidine. The residue was dissolved in pentane (∼2 mL) and the solution was stored at −30 °C to give the final product as yellow solid; yield 59 mg (38%). Anal. calcd for C30H39F9N2OW: C, 45.13; H, 4.92; N, 3.51. Found C, 45.07; H, 4.92; N, 3.52.Synthesis of W(NAr)(CH2CH2)(OTf)2(dme)
A vessel containing a solution of W(NAr)(CHCMe2Ph)(OTf)2(dme) (1.00 g, 1.14 mmol) in 15 mL of toluene was pressurized with ethylene (60 psi) and the reaction mixture was heated to 80 °C for two days. The color of the solution changed from light yellow to orange and an orange solid formed. The microcrystalline solid was filtered off and washed two times with toluene (1 mL); yield 51% (449 mg). Anal. calcd for C20H31F6NO8S2W: C, 30.98; H, 4.03; N, 1.81. Found C, 31.00; H, 3.96; N, 1.70.Reaction of W(NAr)(Ar′NH)(ArNH2)(ORF9) with ethylene
W(NAr)(Ar′NH)(ArNH2)(ORF9) (20 mg, 0.020 mmol), and 1,4-bis-trimethylsilylbenzene (0.15 mL from a stock solution in C6D6; 15 mol%) were dissolved in 0.35 mL of C6D6 in a J. Young NMR tube (1,4-bis-trimethylsilylbenzene was used as an internal standard). Ethylene (15 psi) was added to the tube and the mixture was examined by NMR spectroscopy at room temperature, as described in the text. The tricyclic product lost ethylene readily to reform W(NAr)(Ar′NH)(ArNH2)(ORF9).Synthesis of W(NAr)(py)[(µ-NAr′)(µ-NHAr″)]W(NAr)(ORF9)
A solution of W(NAr)(NHAr′)(ArNH2)(ORF9). (182 mg, 0.190 mmol, 1 equiv.) and cyclohexene (160 mg, 1.9 mmol, 10 equiv.) in 5 mL of C6H6 was heated in an oil bath at 100 °C for 24 h. During that period the color changed from dark red to deep purple. The volatiles were removed in vacuo, and the residue was extracted with pentane. After standing the pentane extract at −30 °C overnight, a crop of orange crystalline starting material was recovered. The pentane was removed in vacuo and pyridine (63 µL, 0.76 mmol, 4 equiv.) and ∼2 mL of ether were added to the residue. The color of the solution changed immediately from deep purple to red and crystals began to form after one hour at room temperature. After storing the mixture at −30 °C overnight, the red crystals were filtered off and washed with cold diethyl ether; yield 77 mg (29%). Anal. calcd for WC56H68F9N5OW2: C, 49.24; H, 5.02; N, 5.13. Found C, 49.43; H, 5.32; N, 5.06.Author contributions
All synthetic work and data analysis were carried out by M. Maji and L. Zeng. SCXRD studies were carried out by V. Carta. R. R. Schrock supervised the project and wrote the original manuscript, and reviewed and edited the manuscript. M. P. Conley supervised, reviewed, and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
CCDC 2419290, 2419291, 2419293, 2419295, 2419296, and 2480801 contain the supplementary crystallographic data for this paper.48a–fData for this article is available from the corresponding authors upon reasonable request. The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: NMR data and spectra, X-ray data, and details of experimental procedures, techniques, and methods. See DOI: https://doi.org/10.1039/d5sc07302b.
Acknowledgements
RRS is grateful to the University of California at Riverside for financial support. MPC thanks the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, Catalysis Science Program, for support through award DE-SC0023344.References
- S. Liu, M. Boudjelel, R. R. Schrock, M. P. Conley and C. Tsay, J. Am. Chem. Soc., 2021, 143, 17209–17218 CrossRef CAS PubMed
.
- M. Boudjelel, R. Riedel, R. R. Schrock, M. P. Conley, A. Berges and V. Carta, J. Am. Chem. Soc., 2022, 144, 10929–10942 CrossRef CAS PubMed
- S. Liu, M. P. Conley and R. R. Schrock, Organometallics, 2022, 41, 1087–1093 CrossRef
- S. Liu, R. R. Schrock, M. P. Conley and V. Carta, Organometallics, 2023, 42, 2321–2325 CrossRef CAS
- S. Liu, R. R. Schrock, M. P. Conley, C. Tsay and V. Carta, Organometallics, 2023, 42, 2251–2261 CrossRef CAS
- R. R. Schrock, R. Riedel, M. Maji, M. Conley and V. Carta, Organometallics, 2023, 42, 2038–2051 CrossRef CAS
- M. Maji, R. Riedel, R. R. Schrock, M. P. Conley and V. Carta, Angew. Chem., Int. Ed., 2024, e202410923 CAS
- M. Maji, A. Sousa-Silva, X. Solans-Monfort, R. R. Schrock, M. P. Conley, P. Farias and V. Carta, J. Am. Chem. Soc., 2024, 146, 18661–18671 CrossRef CAS
- M. Maji, L. Trowbridge, M. Boudjelel, R. R. Schrock, M. P. Conley and V. P. Carta, J. Am. Chem. Soc., 2025, 147, 25462–25470 CrossRef CAS
- J. Rodriguez, M. Boudjelel, L. J. Mueller, R. R. Schrock and M. P. Conley, J. Am. Chem. Soc., 2022, 144, 18761–18765 CrossRef CAS PubMed
-
K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, San Diego, 1997 Search PubMed
- R. R. Schrock and C. Copéret, Organometallics, 2017, 36, 1884–1892 CrossRef CAS
- S. J. McLain, J. Sancho and R. R. Schrock, J. Am. Chem. Soc., 1979, 101, 5451–5453 CrossRef CAS
- B. Blom, H. Clayton, M. Kilkenny and J. R. Moss, Adv. Organomet. Chem., 2006, 54, 149–205 CrossRef CAS
- C. D. Wood, S. J. McLain and R. R. Schrock, J. Am. Chem. Soc., 1979, 101, 3210–3222 CrossRef CAS
- R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CrossRef CAS PubMed
- R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS PubMed
- J. Freundlich, R. R. Schrock, C. C. Cummins and W. M. Davis, J. Am. Chem. Soc., 1994, 116, 6476–6477 CrossRef CAS
- K. F. Hirsekorn, A. S. Veige, M. P. Marchak, Y. Koldobskaya, P. T. Wolczanski, T. R. Cundari and E. B. Lobkovsky, J. Am. Chem. Soc., 2005, 127, 4809–4830 CrossRef CAS
- G. Natta, G. Dall'Asta and G. Mazzanti, Angew. Chem., Int. Ed. Engl., 1964, 3, 723–729 CrossRef
- N. Calderon, E. A. Ofstead, J. P. Ward, W. A. Judy and K. W. Scott, J. Am. Chem. Soc., 1968, 90, 4133–4140 CrossRef CAS
- N. Calderon, J. P. Lawrence and E. A. Ofstead, Adv. Organomet. Chem., 1979, 17, 449 CrossRef CAS
- N. Calderon, Acc. Chem. Res., 1972, 5, 127–132 CrossRef CAS
- D. S. Williams, M.
H. Schofield, J. T. Anhaus and R. R. Schrock, J. Am. Chem. Soc., 1990, 112, 6728–6729 CrossRef CAS
- D. S. Williams, M. H. Schofield and R. R. Schrock, Organometallics, 1993, 12, 4560–4571 CrossRef CAS
- A. Bell, W. Clegg, P. W. Dyer, M. R. J. Elsegood, V. C. Gibson and E. L. Marshall, J. Chem. Soc., Chem. Commun., 1994, 2547–2548 RSC
- A. Bell, W. Clegg, P. W. Dyer, M. R. J. Elsegood, V. C. Gibson and E. L. Marshall, J. Chem. Soc., Chem. Commun., 1994, 2247–2248 RSC
- M. Brookhart, M. L. H. Green and L. Wong, Prog. Inorg. Chem., 1988, 36, 1–124 CAS
- M. Brookhart, M. L. H. Green and G. L. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS
- Y. W. Chao, P. M. Rodgers, D. E. Wigley, S. J. Alexander and A. L. Rheingold, J. Am. Chem. Soc., 1991, 113, 6326–6328 CrossRef CAS
- D. L. Morrison, P. M. Rodgers, Y. W. Chao, M. A. Bruck, C. Grittini, T. L. Tajima, S. J. Alexander, A. L. Rheingold and D. E. Wigley, Organometallics, 1995, 14, 2435–2446 CrossRef CAS
- D. L. Morrison and D. E. Wigley, Inorg. Chem., 1995, 34, 2610–2616 CrossRef CAS
- J. H. Oskam and R. R. Schrock, J. Am. Chem. Soc., 1993, 115, 11831–11845 CrossRef CAS
- L. P. H. Lopez and R. R. Schrock, J. Am. Chem. Soc., 2004, 126, 9526–9527 CrossRef CAS PubMed
- L. P. H. Lopez, R. R. Schrock and P. Müller, Organometallics, 2006, 25, 1978–1986 CrossRef CAS
- R. R. Schrock, J. Chem. Soc., Dalton Trans., 2001, 2541–2550 RSC
- S. C. Marinescu, A. J. King, R. R. Schrock, R. Singh, P. Müller and M. K. Takase, Organometallics, 2010, 29, 6816–6828 CrossRef CAS
- W. C. P. Tsang, K. C. Hultzsch, J. B. Alexander, P. J. Bonitatebus Jr, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 2652–2666 CrossRef CAS
- S. Arndt, R. R. Schrock and P. Müller, Organometallics, 2007, 26, 1279–1290 CrossRef CAS
- R. R. Schrock, L. P. H. Lopez, J. Hafer, R. Singh, A. Sinha and P. Müller, Organometallics, 2005, 24, 5211–5213 CrossRef CAS
- K. S. Lokare, R. J. Staples and A. L. Odom, Organometallics, 2008, 27, 5130–5138 CrossRef CAS
- J. T. Ciszewski, B. Xie, C. Cao and A. L. Odom, Dalton Trans., 2003, 4226–4227 RSC
- C. M. Sever, A. M. Esper, I. Ghiviriga, D. W. Lester, A. Marathianos, C. Ehm and A. S. Veige, ACS Catal., 2025, 15, 5046–5052 CrossRef CAS
- S. S. Nadif, S. A. Gonsales, S. VenkatRamani, I. Ghiviriga, A. S. Veige, T. Kubo, I. Ghiviriga, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2016, 138, 6408–6411 CrossRef CAS PubMed
- S. A. Gonsales, T. Kubo, M. K. Flint, K. A. Abboud, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2016, 138, 4996–4999 CrossRef CAS PubMed
- A. M. Beauchamp, J. Chakraborty, I. Ghiviriga, K. A. Abboud, D. W. Lester and A. S. Veige, J. Am. Chem. Soc., 2023, 145, 22796–22802 CrossRef CAS PubMed
- V. Jakhar, D. Pal, I. Ghiviriga, K. A. Abboud, D. W. Lester, B. S. Sumerlin and A. S. Veige, J. Am. Chem. Soc., 2021, 143, 1235–1246 CrossRef CAS PubMed
-
(a)
CCDC 2419290: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2m6gmy.
| This journal is © The Royal Society of Chemistry 2026 |