
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybridization-tuned dual-chain conjugated polythioether quinones for high-energy rechargeable magnesium batteries
Hongda
Gui
a,
Ze
Wang
a,
Yuliang
Cao
 b and
Fei
Xu
*a
b and
Fei
Xu
*a
aKey Laboratory of Hydraulic Machinery Transients, Ministry of Education, School of Power and Mechanical Engineering, Wuhan University, Wuhan 430072, China. E-mail: xufei2058@whu.edu.cn
bHubei Key Lab of Electrochemical Power Sources, College of Chemistry & Molecular Science, Wuhan University, Wuhan 430072, China
First published on 11th November 2025
Abstract
Organic conjugated polymers featuring abundant redox-active moieties and flexible framework architectures represent promising cathode materials for rechargeable Mg batteries, but reported organic cathodes have struggled to achieve both high reversible capacity and high energy density simultaneously. Herein, three conjugated carbonyl polymers (25PBQS, 26PBQS, and 2356PBQS) are rationally designed by varying the conjugated thioether connectivity patterns of high-capacity, high-voltage benzoquinone units. The benzoquinone units enable high energy density through efficient two-electron redox reactions. The partial sp3 hybridization of sulfur atoms enhances molecular conformational flexibility, facilitating coordination with Mg2+, while partial sp2 hybridization promotes charge delocalization and carbonyl enolization. Compared to single-chain 25PBQS and 26PBQS, dual-chain 2356PBQS with extended π-conjugation demonstrates exceptional active-site utilization (85%), delivering a high reversible capacity of 272 mAh g−1 (50 mA g−1), a high energy density of 339 Wh kg−1 (360 W kg−1), and stable cycling performance with 93.7% capacity retention after 200 cycles. This work provides critical insights into the rational design of organic cathodes with balanced energy and power densities for high-performance rechargeable Mg batteries.
1. Introduction
Li-ion batteries (LIBs), with their high energy density and long lifespan, currently dominate mobile energy-storage markets, including electric vehicles and consumer electronics.1,2 However, their application in grid energy storage faces inherent limitations due to finite Li reserves and safety concerns (e.g., dendrite-induced thermal runaway).3 In principle, rechargeable Mg batteries (RMBs) emerge as a suitable alternative for large-scale energy storage. The Mg metal anode offers advantages including high crustal abundance (480 times higher than Li), low redox potential (−2.37 V vs. SHE), high theoretical capacity (2205 mAh g−1 and 3833 mAh cm−3), and negligible dendritic growth during cycling, ensuring intrinsic safety.4 Over the past decade, breakthroughs in Mg-compatible electrolytes (particularly the non-nucleophilic Mg[B(hfip)4]2/G2-DME electrolyte) and scalable Mg salt synthesis have laid a robust foundation for RMB development.5–7 Nevertheless, the exploration of high-performance cathode materials lags significantly, forming a critical bottleneck to advancing RMB technologies.Compared to monovalent alkali metal cations, the high charge density and strong polarization effects of divalent Mg2+ lead to sluggish solid-state diffusion kinetics and limited active sites for Mg storage in inorganic lattices.8 Conventional inorganic Mg-storage cathodes suffer from structural inflexibility, low reversible capacity, and intrinsic kinetic barriers.9 In contrast, organic conjugated polymers offer resource-abundant, structurally tunable selections for Mg-storage cathodes. Their open and flexible frameworks exhibit low cation-selectivity constraints, while the extended π-conjugation effectively delocalizes the negative charge to mitigate strong Mg2+–host interactions, which are critical advantages for high-performance Mg-storage cathodes.10 Although research on organic RMB cathodes remains nascent, reported polymer cathodes (e.g., polyimides and quinone-based polymers) already demonstrate superior Mg-storage capacities and rapid reaction kinetics.10–12 Organic RMBs, integrating polymer cathodes with metallic Mg anodes, align with sustainability and safety requirements, making them an attractive direction for next-generation grid-scale energy-storage technologies.
Organic conjugated polymers typically comprise electroactive units interconnected via linker groups. Among various electroactive units, benzoquinone derivatives emerge as promising candidates due to their high theoretical specific capacity (496 mAh g−1) and low LUMO energy level favoring high energy densities. From the perspective of linker design, incorporation of sulfur atoms between benzoquinone units to construct conjugated thioether architectures (C–S–C) offers advantages because of their mixed sp3 and sp2 hybridization states. Specifically, the sp3 hybridization of sulfur atoms enhances conformational flexibility, thereby promoting effective Mg2+ coordination with carbonyl groups. On the other hand, the sp2-hybridized component enables p–π conjugation, while the inherent electronegativity of sulfur further modulates the LUMO energy level, synergistically improving redox potentials.13,14 Guided by these principles, three conjugated polythioether benzoquinone polymers (denoted as 25PBQS, 26PBQS, and 2356PBQS; molecular structures and theoretical capacities are shown in Fig. 1a) are developed in the present study. These polymers demonstrate exceptional Mg-storage performance, with high discharge plateaus approaching 2.0 V vs. Mg2+/Mg and remarkable specific capacities over 250 mAh g−1 at 50 mA g−1, as well as superior rate capabilities. The corresponding energy/power densities reach 351 Wh kg−1/7015 W kg−1 for 25PBQS, 314.5 Wh kg−1/6485 W kg−1 for 26PBQS, and 402 Wh kg−1/6177 W kg−1 for 2356PBQS, which are of the highest level among the reported RMB cathodes. Notably, 2356PBQS maintains a high energy density of 360 Wh kg−1 at an elevated power density of 339 W kg−1, surpassing most organic cathode materials for Mg batteries. Mechanistic investigations and theoretical calculations reveal that the dual-thioether chain in 2356PBQS enhances electron delocalization and promotes carbonyl enolization, resulting in its superior performance compared to single-chain analogues (25PBQS and 26PBQS). This work provides critical insights into the rational design of organic cathodes with balanced energy and power densities for next-generation RMBs.
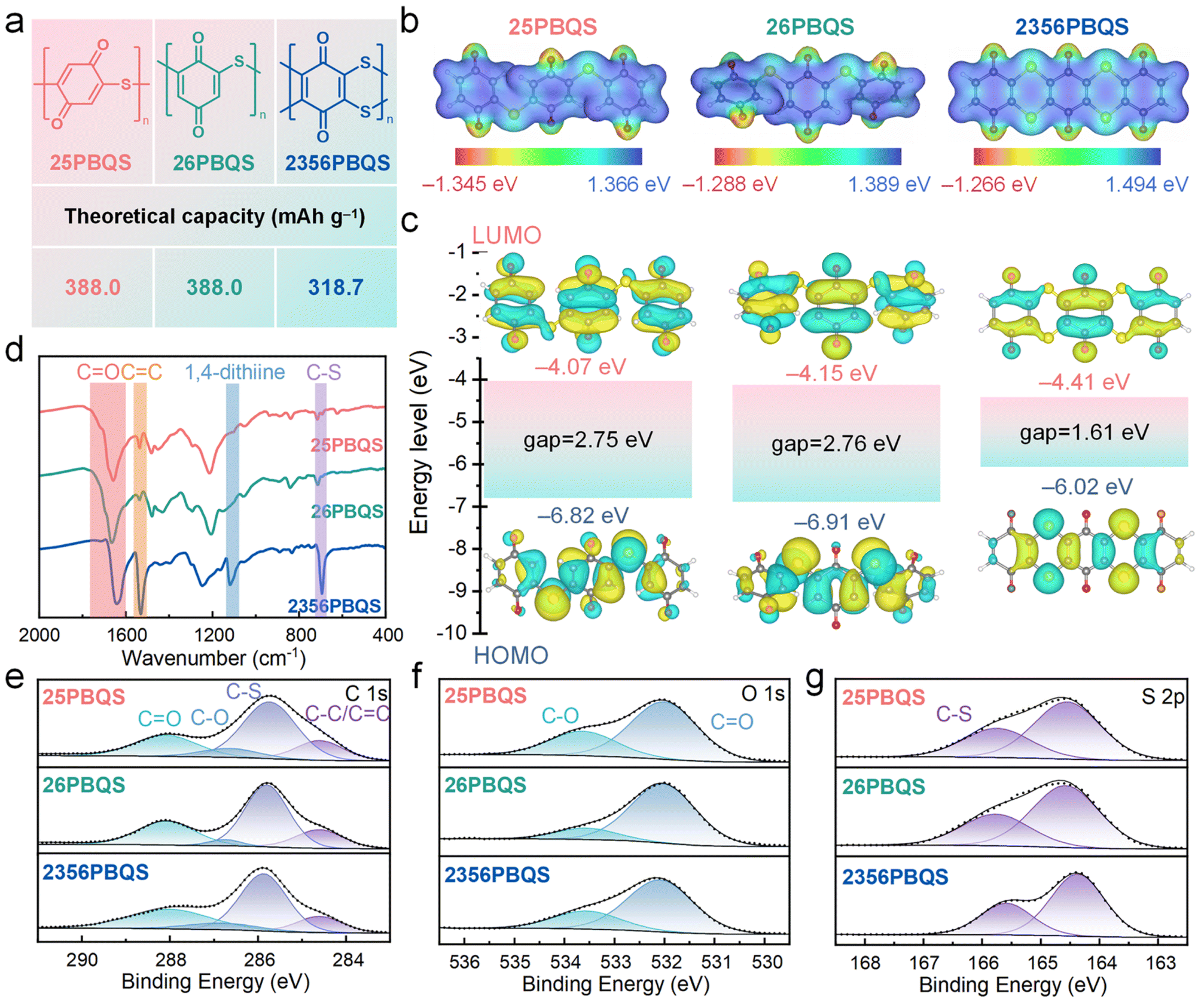 | ||
| Fig. 1 (a) Structures and theoretical capacities, (b) ESP surfaces, (c) HOMO–LUMO energy level diagrams, (d) FTIR spectra, and (e) C 1s, (f) O 1s and (g) S 2p XPS spectra of 25PBQS, 26PBQS and 2356PBQS. | ||
2. Experimental
2.1 Synthesis of 25PBQS
Na2S·9H2O (5.5 mmol, 99.99%, Aladdin) was ground into a powder and added to 20 mL of N-methyl-2-pyrrolidone (NMP, 99%, Sinopharm Chemical Reagent Co., Ltd). After sonication for 15 minutes, 2,5-dichloro-1,4-benzoquinone (5 mmol, 97%, Bide Pharmatech Ltd) was introduced, and the mixture was stirred for 45 minutes. The reaction proceeded at 160 °C for 24 hours. The resulting mixture was diluted with 60 mL of deionized water, and concentrated hydrochloric acid (HCl, 36.0–38.0%, Sinopharm Chemical Reagent Co., Ltd) was added dropwise to adjust the pH to 1–2. The mixture was filtered, and the precipitate was washed with deionized water (three times). For further purification, the precipitate was dissolved in 40 mL of NaOH solution (0.25 M), and hydrochloric acid was added dropwise to reprecipitate the product at pH 1–2. The mixture was centrifuged, washed with hydrochloric acid (0.1 M, four times) and ethanol containing 0.1 M HCl (once), then dried under airflow at 80 °C for 6 hours and vacuum-dried at 60 °C for 12 hours. The resulting product was ground into a powder, which was then treated with carbon disulfide (CS2, 99.7%, Sinopharm Chemical Reagent Co., Ltd) for 1 hour, centrifuged, and vacuum-dried again at 60 °C for 12 hours to yield 304 mg of a black solid (44% yield). To oxidize phenolic groups to carbonyls, the intermediate (304 mg) was dispersed in 40 mL of tetrahydrofuran (THF, 99.5%, Sinopharm Chemical Reagent Co., Ltd) via sonication. Two equivalents of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 98%, Aladdin) were added, and the mixture was stirred at room temperature for 3 days. The product was centrifuged, washed with THF (four times), and vacuum-dried at 60 °C overnight to obtain 244 mg of 25PBQS as a black solid (yield: 81%).2.2 Synthesis of 26PBQS
A procedure analogous to that described above was employed using 2,6-dichloro-1,4-benzoquinone (98%, Bide Pharmatech Ltd) as the starting material. The intermediate product (400 mg, yield: 58%) was obtained as a black solid, which was oxidized to yield 302 mg of 26PBQS as a black solid (yield: 76%).2.3 Synthesis of 2356PBQS
Following a similar protocol, tetrachloro-1,4-benzoquinone (5 mmol, 98%, Aladdin) was reacted with Na2S·9H2O (11 mmol) in NMP (30 mL). The intermediate product (626 mg, yield: 75%) was isolated as a yellow-brown solid. Oxidation yielded 561 mg of 2356PBQS as a black solid (yield: 94%).2.4 Structural characterization studies
Fourier transform infrared (FTIR) spectroscopy was performed using a Nicolet iS50 FTIR spectrometer (Thermo Fisher Scientific), with micro-FTIR analysis conducted on a Nicolet iN10 microscope (Thermo Fisher Scientific). X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB Xi+ system. X-ray diffraction (XRD) patterns were collected on a Rigaku Ultima IV diffractometer, and thermogravimetric analysis (TGA) was conducted on a Mettler-Toledo TGA2 instrument. Morphological analysis and elemental mapping were carried out using field-emission scanning electron microscopy (FESEM, MIRA3LMH) and transmission electron microscopy (TEM, JEM-2100) coupled with EDS. Nitrogen adsorption isotherms were measured at 77 K using liquid nitrogen with a Micromeritics ASAP 2460 instrument. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) model. Prior to ex situ characterization, electrodes were rinsed with dehydrated 1,2-dimethoxyethane (DME) to remove residual electrolytes.2.5 Electrochemical measurements
Cathodes were prepared by ball-milling active materials with Ketjenblack (KB) at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio (250 rpm, 2 hours), followed by mixing with polytetrafluoroethylene (PTFE) binder (9:1 mass ratio) in 2-propanol. The slurry was coated onto carbon paper and vacuum-dried at 60 °C for 12 hours. Electrodes (12 mm diameter and 1.5 ± 0.3 mg cm−2 active mass loading) were assembled into CR2032 coin cells using polished magnesium foil as the anode, Whatman GF/A separators, and molybdenum spacers at the cathode side to prevent corrosion. Two electrolytes were used: (1) a chloride-containing electrolyte comprising 0.25 M Mg(TFSI)2 and 0.48 M MgCl2 in DME and (2) a chloride-free electrolyte comprising 0.5 M (Mg(DME)3)(B(hfip)4)2 in diglyme (G2), synthesized via a literature method.7 Cycling performance was evaluated using the galvanostatic intermittent titration technique (GITT) on a LAND CT3002A system, while cyclic voltammetry (CV) was performed on a CHI660e electrochemical workstation.
:1 mass ratio (250 rpm, 2 hours), followed by mixing with polytetrafluoroethylene (PTFE) binder (9:1 mass ratio) in 2-propanol. The slurry was coated onto carbon paper and vacuum-dried at 60 °C for 12 hours. Electrodes (12 mm diameter and 1.5 ± 0.3 mg cm−2 active mass loading) were assembled into CR2032 coin cells using polished magnesium foil as the anode, Whatman GF/A separators, and molybdenum spacers at the cathode side to prevent corrosion. Two electrolytes were used: (1) a chloride-containing electrolyte comprising 0.25 M Mg(TFSI)2 and 0.48 M MgCl2 in DME and (2) a chloride-free electrolyte comprising 0.5 M (Mg(DME)3)(B(hfip)4)2 in diglyme (G2), synthesized via a literature method.7 Cycling performance was evaluated using the galvanostatic intermittent titration technique (GITT) on a LAND CT3002A system, while cyclic voltammetry (CV) was performed on a CHI660e electrochemical workstation.
2.6 Theoretical computations
All density functional theory (DFT) calculations were conducted using Gaussian 16.15 25PBQS, 26PBQS and 2356PBQS were optimized at the B3LYP/6-311G(d,p) level, while anionic and Mg-coordinated systems were treated with the UB3LYP functional and 6-31+G(d,p) basis set for geometry optimization, followed by single-point energy calculations at the 6-311+G(d,p) level.16,17 Dispersion corrections were applied using the DFT-D3 method with Becke–Johnson damping, and solvent effects (DME) were modeled via the IEFPCM implicit solvent model.18–21 Wavefunction stability was verified for all systems. Post-processing analyses, including orbital energy evaluations, electrostatic potential (ESP),22 electron localization function (ELF),23 Harmonic Oscillator Model of Aromaticity (HOMA) indices,24 Localized Orbital Locator-π (LOL-π),25,26 Atomic Dipole Moment Corrected Hirshfeld (ADCH),27 fuzzy bond orders,28 and Reduced Density Gradient (RDG),29 were performed using Multiwfn software.30,31 Visualization of RDG utilized VMD,32 while structural representations were generated with VESTA.333. Results and discussion
Three representative polythioether benzoquinone polymers (25PBQS, 26PBQS, and 2356PBQS; Fig. 1a) were synthesized and investigated comparatively. As illustrated in Fig. 1a, 25PBQS and 26PBQS exhibit single-chain structures, whereas 2356PBQS features a dual-chain architecture. While 2356PBQS has maximized p–π conjugation and the lowest theoretical capacity (318.7 mAh g−1, calculated solely based on C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups as redox-active sites), subsequent electrochemical testing unexpectedly reveals its superior practical Mg-storage capacity compared to its single-chain analogues. Fig. 1b shows the ESP maps of 25PBQS, 26PBQS, and 2356PBQS (trimer models). Notably, 2356PBQS exhibits coplanar benzoquinone units owing to its dual sulfur chain structure, a distinct contrast to the non-coplanar configurations of 25PBQS and 26PBQS. This coplanar structure promotes sp2 hybridization of the sulfur atoms and facilitates extensive p–π conjugation along the polymer backbone, thereby enhancing charge delocalization and structural integrity. Further insight from ELF isosurfaces (Table S2) reveals that in 2356PBQS, the p-orbitals of sulfur are oriented perpendicular to the molecular plane, unlike those in 25PBQS and 26PBQS. This distinct alignment signifies a greater degree of sp2-hybridized character and enhanced p–π conjugation within the dual sulfur chain framework of 2356PBQS, working synergistically to improve electron delocalization. The low ESP regions at oxygen sites (Fig. 1b) indicate their high tendency for Mg2+ coordination. HOMO–LUMO distributions (Fig. 1c) and energy levels further predict performance trends. All polymers exhibit LUMO primarily on benzoquinone units, with partial delocalization onto C–S bonds. This is a consequence of partial sp2 hybridization of the sulfur atoms and p–π conjugation, enhancing electron-withdrawing capability. The low LUMO energies (−4.41 to −4.07 eV) preserve the high redox potential of benzoquinone, critical for improved energy density. 2356PBQS has the lowest LUMO energy (−4.41 eV) due to the electronegativity of sulfur and dual-chain-enabled coplanarity, which enhances conjugation and charge delocalization. It is noteworthy that, 2356PBQS's narrow HOMO–LUMO gap (1.54 eV vs. 2.64 eV for 25PBQS and 2.66 eV for 26PBQS) signifies enhanced electronic conductivity, directly originating from its extended dual-chain p–π conjugation.34
O groups as redox-active sites), subsequent electrochemical testing unexpectedly reveals its superior practical Mg-storage capacity compared to its single-chain analogues. Fig. 1b shows the ESP maps of 25PBQS, 26PBQS, and 2356PBQS (trimer models). Notably, 2356PBQS exhibits coplanar benzoquinone units owing to its dual sulfur chain structure, a distinct contrast to the non-coplanar configurations of 25PBQS and 26PBQS. This coplanar structure promotes sp2 hybridization of the sulfur atoms and facilitates extensive p–π conjugation along the polymer backbone, thereby enhancing charge delocalization and structural integrity. Further insight from ELF isosurfaces (Table S2) reveals that in 2356PBQS, the p-orbitals of sulfur are oriented perpendicular to the molecular plane, unlike those in 25PBQS and 26PBQS. This distinct alignment signifies a greater degree of sp2-hybridized character and enhanced p–π conjugation within the dual sulfur chain framework of 2356PBQS, working synergistically to improve electron delocalization. The low ESP regions at oxygen sites (Fig. 1b) indicate their high tendency for Mg2+ coordination. HOMO–LUMO distributions (Fig. 1c) and energy levels further predict performance trends. All polymers exhibit LUMO primarily on benzoquinone units, with partial delocalization onto C–S bonds. This is a consequence of partial sp2 hybridization of the sulfur atoms and p–π conjugation, enhancing electron-withdrawing capability. The low LUMO energies (−4.41 to −4.07 eV) preserve the high redox potential of benzoquinone, critical for improved energy density. 2356PBQS has the lowest LUMO energy (−4.41 eV) due to the electronegativity of sulfur and dual-chain-enabled coplanarity, which enhances conjugation and charge delocalization. It is noteworthy that, 2356PBQS's narrow HOMO–LUMO gap (1.54 eV vs. 2.64 eV for 25PBQS and 2.66 eV for 26PBQS) signifies enhanced electronic conductivity, directly originating from its extended dual-chain p–π conjugation.34
The three PBQSs were synthesized via nucleophilic substitution reactions (Fig. S1), and the amorphous structures are evidenced by broad XRD peaks distinct from precursor materials (Fig. S3). The FTIR spectra of the three polymers (Fig. 1d) share similarities due to their analogous chemical frameworks. However, 2356PBQS displays a distinct sharp peak at 1119 cm−1, characteristic of its unique 1,4-dithiine structure (dual sulfur chain). The strong CO stretching vibrations are observed at 1658 (25PBQS), 1666 (26PBQS), and 1644 cm−1 (2356PBQS). The redshift in the CO vibration frequency of 2356PBQS arises from conjugation with the adjacent 1,4-dithiine, reflecting enhanced electron delocalization.35–38 Peaks at 1536 (25PBQS), 1537 (26PBQS), and 1531 cm−1 (2356PBQS) correspond to CC vibrations, while C–S stretching modes appear at 715 (25PBQS), 714 (26PBQS), and 694 cm−1 (2356PBQS), confirming sulfur incorporation.39,40 The redshift and enhancement of C–S absorption in 2356PBQS further corroborate extended π-electron delocalization due to its expanded conjugation.
Elemental analysis (Fig. S5) estimates polymerization degrees of about 37, 33, and 42 for 25PBQS, 26PBQS, and 2356PBQS, respectively, based on S/Cl ratios (18.1, 16.1, and 20.5, respectively), indicating the suppressed dissolution in electrolytes and enhanced cycling stability. The C 1s, O 1s, and S 2p XPS spectra are shown in Fig. 1e–g. The C 1s spectra (Fig. 1e) exhibit characteristic peaks at 284.6 (C–C/CC), 285.8 (C–S), 286.8 (C–O) and 288.0 eV (CO).41 In the O 1s spectra (Fig. 1f), peaks at 532.1 (CO) and 533.6 eV (C–O) are observed, with the latter attributed to partial surface reduction of CO groups during synthesis (see the experimental details for discussion). The S 2p spectra of 25PBQS and 2356PBQS (Fig. 1g) display a doublet at 164.4 (2p3/2) and 165.6 eV (2p1/2), confirming successful sulfur incorporation via C–S bond formation.41 Due to the slightly lower p–π conjugation degree of sulfur in 26PBQS (meta-), the S 2p spectrum shows a dual state at the positions with slightly higher binding energy (164.6 (2p3/2) and 165.8 eV (2p1/2)). TGA profiles under N2 exhibit similar decomposition temperatures (∼295 °C) for all PBQSs, confirming the structural similarity (Fig. S7). These results collectively validate the successful synthesis of high-polymerization-degree, sulfur-linked benzoquinone polymers with tailored electronic properties for Mg storage.
While previous studies have indicated that organic RMB cathodes generally suffer from relatively low operating voltages, the galvanostatic charge–discharge profiles at 100 mA g−1 (Fig. 2a–c) and CV curves (Fig. 2d, after activation, see Fig. S8 for the other cycles) of 25PBQS, 26PBQS, and 2356PBQS demonstrate their advantages. Their respective midpoint discharge voltages are 1.82 V (25PBQS), 1.79 V (26PBQS), and 1.91 V (2356PBQS). The CV curves of 25PBQS, 26PBQS and 2356PBQS exhibit reduction peaks at notably high potentials of 1.98 V, 1.88 V, and 2.06 V vs. Mg2+/Mg, respectively (Fig. 2d and S8), outperforming those of most reported organic RMB cathodes (comparison data in Table S3). Such high operating voltages directly contribute to enhanced energy density, consistent with their low-lying LUMO energy levels (Fig. 1c). 2356PBQS demonstrates the highest reduction potential (2.06 V), aligning with its lowest LUMO energy (−4.41 eV). All three cathodes undergo a characteristic activation process during cycling (Fig. 2e and S9), during which the capacity gradually increases and polarization decreases as the polymer chains adopt optimized conformations for improved coordination with the bivalent Mg2+ cations.42 The activation rates differ among the three cathodes, likely due to their distinct molecular structures. The single-chain structures of 25PBQS and 26PBQS allow faster activation due to greater conformational flexibility, whereas the rigid dual-chain architecture of 2356PBQS slows this process. Despite having a lower theoretical capacity, 2356PBQS delivers the highest practical capacity of 247 mAh g−1, exceeding that of 25PBQS (218 mAh g−1) and 26PBQS (194 mAh g−1). This paradox will be mechanistically explained in the following sections.
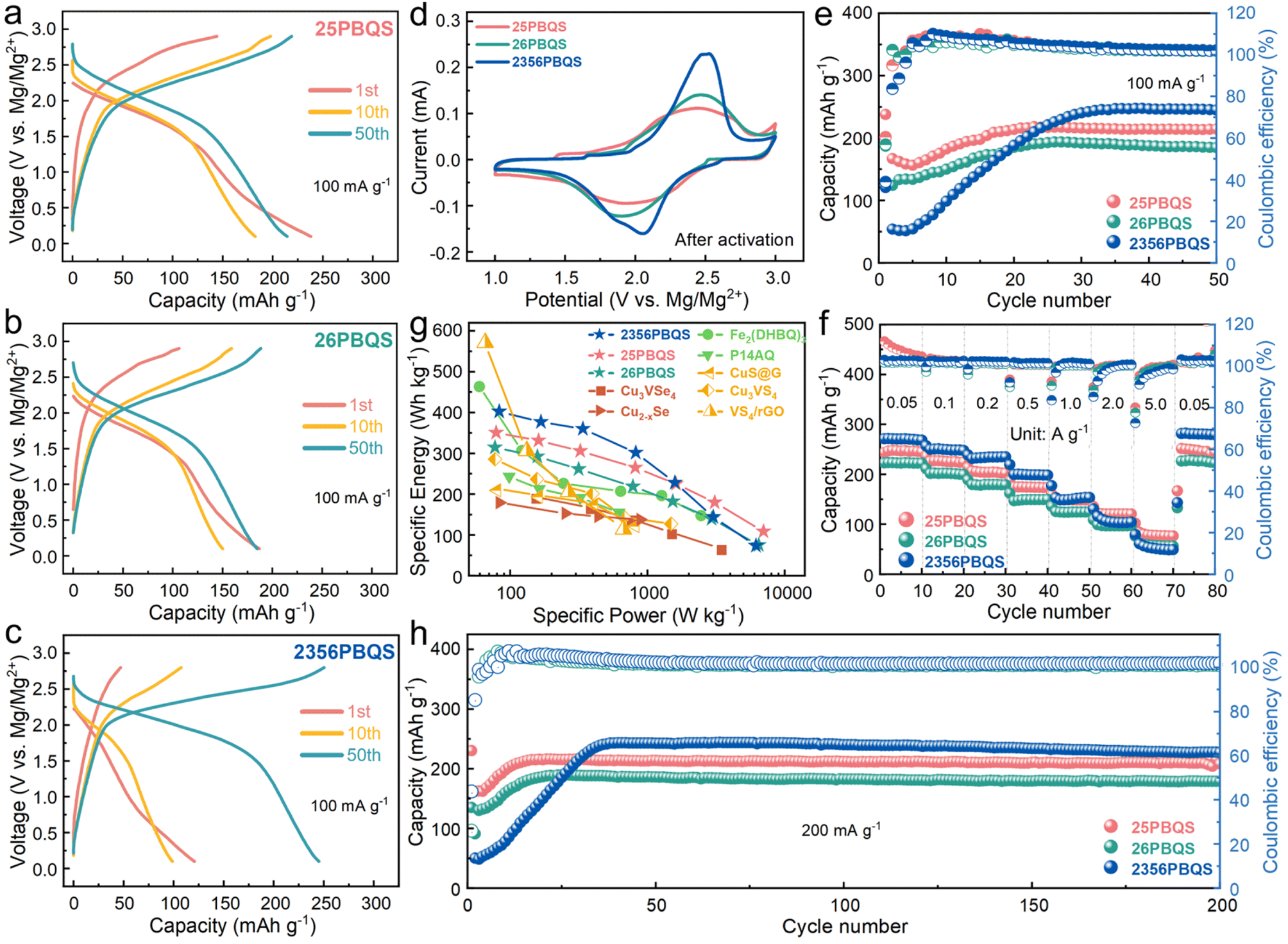 | ||
| Fig. 2 (a–c) Charge–discharge profiles (100 mA g−1), (d) typical CV curves (0.1 mV s−1), (e) cycling performances (100 mA g−1), (f) rate performance (after activation) and (h) long-term cycling performance (200 mA g−1) of 25PBQS, 26PBQS, and 2356PBQS. (g) Comparison of specific energy densities of high-performance RMB cathode materials (Cu3VSe4,43 Cu2–xSe,8 Fe2(DHBQ)3,44 P14AQ,45 CuS@G,46 Cu3VS4 (ref. 47) and VS4/rGO48). | ||
Benefiting from their open polymer frameworks and delocalized negative charges, these PBQS cathodes demonstrate exceptional rate performance, with 2356PBQS delivering superior capacities of 272, 251, 235, 201, 154, 101, and 61 mAh g−1 at current densities ranging from 50 to 5000 mA g−1, exceeding those of 25PBQS and 26PBQS (Fig. 2f; charge/discharge profiles in Fig. S11). Ragone plot comparisons (Fig. 2g) further highlight their leading position among state-of-the-art RMB cathodes (details in Tables S4–S6), where 25PBQS (350.6 Wh kg−1, 7015 W kg−1), 26PBQS (314.5 Wh kg−1, 6485 W kg−1), and notably 2356PBQS (402.3 Wh kg−1, 6176.8 W kg−1) achieve record energy and power densities owing to their high operating voltages, capacities, and rate capability. Remarkably, 2356PBQS maintains 360.2 W kg−1 at 339.1 Wh kg−1, representing the highest combined performance reported for RMB cathodes. Long-term cycling at 200 mA g−1 (Fig. 2h) confirms excellent stability, with retention rates of 96.4%, 94.7%, and 93.7% after 200 cycles for 25PBQS (216 mAh g−1), 26PBQS (189 mAh g−1), and 2356PBQS (243 mAh g−1), respectively. Overall, with superior rate performance and exceptional energy and power densities, 25PBQS and 2356PBQS emerge as comprehensively high-performance organic cathode materials for RMBs.
The open frameworks of organic materials necessitate identification of the charge carriers (Mg2+ or MgCl+).45,49 Electrochemical tests in chloride-containing electrolytes across a range of concentrations (Fig. S13a–c) show minimal capacity variation (<10%) after activation, indicating negligible dependence of Mg storage on the Cl− concentration. Furthermore, in a chloride-free Mg[B(hfip)4]2/G2-DME electrolyte (electrolyte characterization in Fig. S14), the polymers deliver reversible capacities of 179 (25PBQS), 148 (26PBQS) and 184 mAh g−1 (2356PBQS) at 200 mA g−1 (Fig. 3a; additional cycling, rate performance, and Mg2+ diffusion data in Fig. S15–S18). Despite slightly reduced cycling stability in the chloride-free electrolyte (attributed to discharge product dissolution, Fig. S19), the capacity trends mirror those in chloride-containing electrolyte (216, 185, and 227 mAh g−1), with chloride-free capacities reaching about 80% of their chloride-containing counterparts (Fig. S20). TEM-EDS analysis of 2356PBQS at the 35th cycle in chloride-containing electrolyte (Fig. 3b–d) quantified a clear difference in Mg content between discharged (16.00%) and charged states (9.60%), correlating directly with magnesiation and demagnesiation. The residual Mg in the charged state is attributed to minor irreversibility and possible electrolyte retention. Although Cl content increased from 1.50% (charged) to 2.30% (discharged), this 0.80% change is negligible compared to the 6.40% shift in Mg, further confirming that Mg2+ is the primary stored cation (Table S7; carbon excluded due to interference). These results consistently demonstrate a Mg2+-dominated storage mechanism across all three polymers.
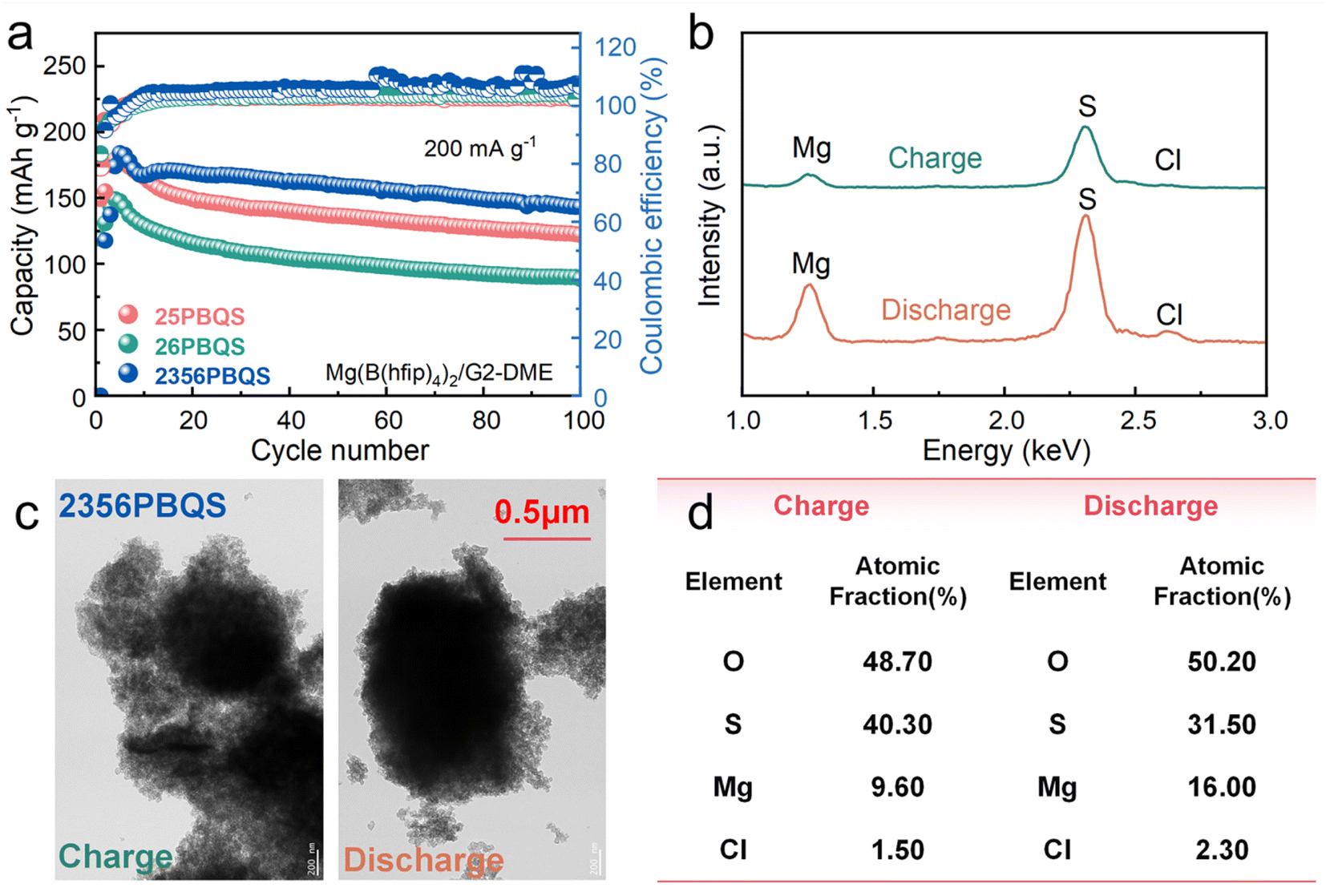 | ||
| Fig. 3 (a) Cycling performances of 25PBQS, 26PBQS and 2356PBQS in Cl-free Mg(B(hfip))4/G2-DME electrolyte. (b) EDS spectra, (c) TEM images and (d) elemental compositions of 2356PBQS at the charge/discharge states of the 35th cycle in Cl-containing Mg(TFSI)2-MgCl2/DME electrolyte. | ||
To elucidate the redox mechanism, ex situ FTIR analyses were conducted on 25PBQS, 26PBQS and 2356PBQS during cycling in Mg(TFSI)2-MgCl2/DME electrolyte (Fig. 4a–c and S21). All three polymers exhibit reversible carbonyl enolization, evidenced by diminished CO peaks in the discharged states and their recovery upon charging. The CO stretching bands redshift during discharge, reflecting reduced double-bond character and enhanced conjugation.50,51 Discharged states display new aromatic skeleton vibrations (1560, 1540, 1510 and 1480 cm−1) and S–Ar stretching modes (∼1050 cm−1), consistent with quinoid-to-benzenoid transitions during reduction.13 Notably, 2356PBQS exhibits unique structural change features. Its characteristic 1,4-dithiine peak (1119 cm−1) vanishes completely upon discharge, signifying conjugation enhancement within the sulfur-bridged framework. Furthermore, the CO band of 2356PBQS redshifts to 1632 cm−1 (discharged), markedly lower than 25PBQS (1647 cm−1) and 26PBQS (1647/1636 cm−1), highlighting dual sulfur chain-enabled conjugation extension and charge delocalization. These structural advantages account for the higher practical capacity of 2356PBQS (272 mAh g−1) despite its lower theoretical value (318.7 mAh g−1), as conjugated electron delocalization stabilizes the enolized structure and improves redox reversibility.
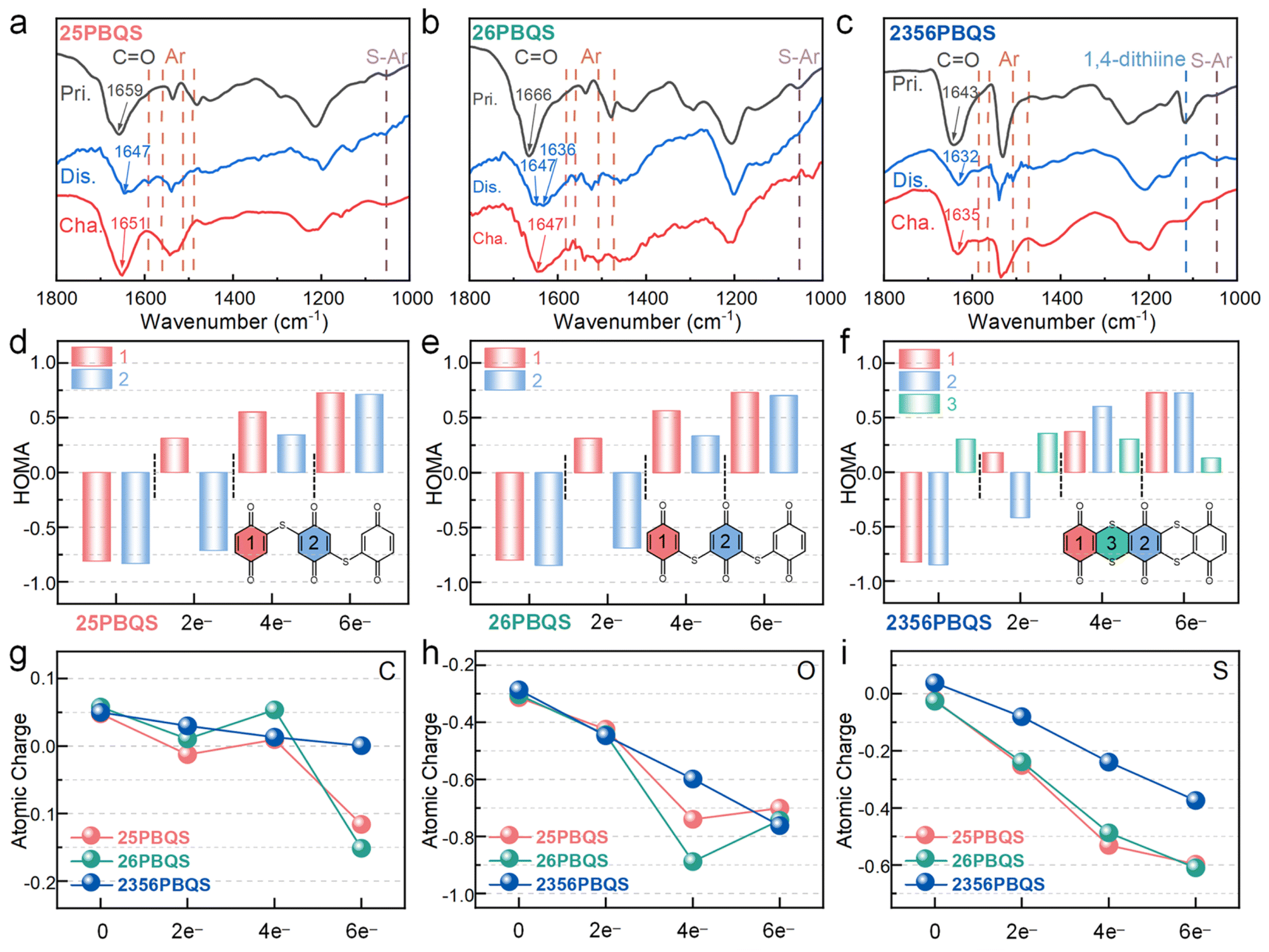 | ||
| Fig. 4 FTIR spectra of (a) 25PBQS, (b) 26PBQS, and (c) 2356PBQS in Mg(TFSI)2-MgCl2/DME electrolyte. HOMA indices of (d) 25PBQS, (e) 26PBQS, and (f) 2356PBQS. Atomic charges of (g) C, (h) O, and (i) S atoms. | ||
DFT calculations were employed to evaluate the conjugation evolution of 25PBQS, 26PBQS, and 2356PBQS across reduction states (Fig. 4d–f). The HOMA indices reveal aromaticity trends. In the pristine state, benzoquinone rings (rings 1 and 2) exhibit negative HOMA values (antiaromaticity, 4 π-electrons), while the 1,4-dithiine of 2356PBQS (ring 3) shows a positive HOMA value (aromaticity), confirming p–π conjugation via the partial sp2 hybridization of sulfur. Upon reduction, all rings gradually adopt positive HOMA indices, indicating enhanced aromaticity and π-electron delocalization. Significantly, the 1,4-dithiine of 2356PBQS maintains positive HOMA throughout reduction, unlike single-chain analogs (25/26PBQS), highlighting dual sulfur chain-enabled conjugation enhancement. LOL-π analyses further quantify π-electron delocalization (Table S8). In contrast to 25PBQS and 26PBQS exhibiting fragmented π-electron distributions in reduced states, 2356PBQS displays continuous LOL-π isosurfaces, confirming superior electron delocalization. Fig. 4g–i depict the atomic charge evolution of C, O and S atoms in 25PBQS, 26PBQS and 2356PBQS during redox processes, calculated via the ADCH method. During electron uptake, 25PBQS and 26PBQS exhibit a nonlinear charge redistribution on C and O atoms, marked by anomalous behavior in the 4-electron and 6-electron reduced states. Transitioning from the 2-electron to 4-electron state, C atoms lose electron density, while O atoms undergo abrupt accumulation. Conversely, advancing to the 6-electron state, C atoms rapidly regain electron density as electrons migrate from O to C atoms. This underscores the inferior electron delocalization of 25PBQS4− and 26PBQS4− compared to 2356PBQS. In contrast, 2356PBQS shows near-linear charge evolution across C, O, and S atoms during reduction, reflecting sustained electron delocalization. Notably, sulfur in 2356PBQS adopts a positive charge in the oxidized state (vs. negative charge in 25/26PBQS), directly evidencing enhanced p–π conjugation with CC via its dual sulfur chain architecture. These results indicate that the extended conjugation framework of 2356PBQS stabilizes the electron distribution, enabling superior redox reversibility and a high practical capacity.
To further elucidate the redox mechanism, XPS analysis was performed on 2356PBQS in various charge/discharge states (Fig. 5a–c, S22 and S23; XPS spectra of 25PBQS and 26PBQS in Fig. S24). In the C 1s spectra (Fig. 5a), the dominant C–C peak at 284.6 eV from conductive carbon complicates direct quantification of C–O (286.9 eV) and CO (288.6 eV) intensity changes, but the appearance of a π–π* satellite peak at 293.2 eV confirms enhanced electron delocalization within the conjugated framework during reduction. The differing 2356PBQS peak intensities in C 1s spectra may originate from either precipitation of dissolved 2356PBQS–Mg complexes or outward material migration during cycling (Fig. S19). The O 1s spectra (Fig. 5b) exhibit reversible intensity variations of CO and C–O, consistent with FTIR results and support the carbonyl enolization mechanism. A new C–O–Mg peak emerges at 532.1 eV in the discharged state, which is absent in monovalent cation storage systems, resulting from Mg2+ coordination to enolized oxygen, attributable to its high charge density.42 Additionally, a new SO peak is observed at 532.8 eV in both charged and discharged states, exclusively in Mg(TFSI)2-MgCl2/DME electrolyte,52 corroborated by S 2p spectra (Fig. 5c) where SO bonds (169.5 and 170.6 eV) are detected but diminishing with prolonged electrode washing time (Fig. S22). These SO features, absent in Mg[B(hfip)4]2/G2-DME electrolyte, are attributed to TFSI− adsorption and/or decomposition.53 A −0.5 eV shift in C–S binding energy is observed during discharge, indicative of increased electron density around sulfur atoms due to charge delocalization. In the pristine state, sulfur is bonded to CC units in a configuration resembling sp2 hybridization. Upon discharge, electrons populate the conduction band and become delocalized throughout the conjugated framework, promoting a shift toward sp3 hybridization. This increase in electron density at sulfur sites leads to a corresponding shift of the binding energy to lower values. During charging, electron extraction partially restores the initial electron distribution, resulting in a return of the peak position toward its original energy.
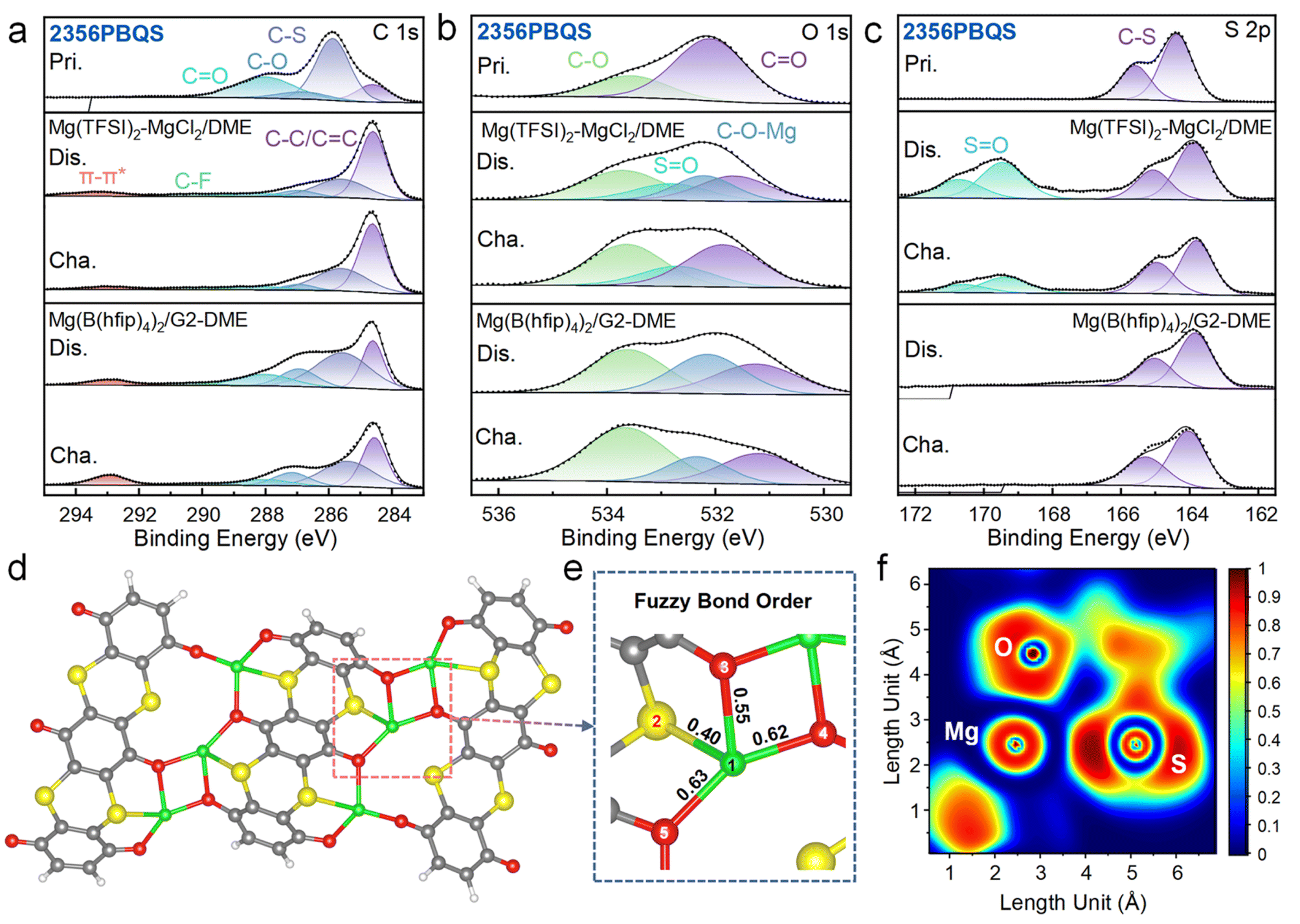 | ||
| Fig. 5 (a) C 1s, (b) O 1s, and (c) S 2p XPS spectra of 2356PBQS in the pristine and charge/discharge states in Mg(TFSI)2-MgCl2/DME and Mg(B(hfip)4)2/G2-DME electrolytes. (d) Molecular structure, (e) partial view with fuzzy bond orders and (f) ELF of 2356PBQS–Mg (Mg1, S2 and O5). | ||
DFT simulations further elucidate the Mg2+ coordination features (Fig. 5d). Owing to the large separation (5.30 Å) between CO groups within a single 2356PBQS chain, Mg2+ preferentially bridges CO sites from adjacent chains. Structural optimization of a model consisting of three 2356PBQS chains, six Mg2+ ions, and twelve electrons reveals chain bending, enabled by sp3 hybridization of sulfur, facilitating multi-O atom coordination. Fuzzy bond order analysis, which quantifies delocalization and bond strength through continuous values (overcoming the limitations of classical integer bond orders), indicates a weak interaction between Mg2+ and S (Mg1–S2 = 0.40; Fig. 5e). ELF shows polarization-induced distortion around Mg2+ and a decrease in sulfur charge from −0.374 to 0.034 upon coordination, indicating electron attraction by Mg2+. This attractive interaction is further supported by RDG analysis (Fig. S29), suggesting a weak interaction between S and Mg2+.
4. Conclusion
In summary, three polythioether benzoquinone polymers, 25PBQS, 26PBQS, and 2356PBQS were synthesized and studied comparatively as RMB cathodes. Dual-sulfur-chain 2356PBQS, with a theoretical capacity of 319 mAh g−1, exhibited a higher reversible capacity (272 mAh g−1 at 50 mA g−1) compared to single-chain 25PBQS and 26PBQS (theoretical capacity of 388 mAh g−1 and reversible capacities of 247 and 223 mAh g−1 at 50 mA g−1, respectively). This performance originates from the partial sp2 hybridization of sulfur atoms and dual-sulfur chain, which improves charge delocalization and structural stability, as validated by spectroscopic and computational analyses. Mechanistic investigation revealed that all three polymers undergo carbonyl enolization, but 2356PBQS uses its extended conjugation structure to achieve improved charge delocalization and enhanced redox reversibility. Atomic charge analysis confirmed stronger electron delocalization in 2356PBQS, while its sp3-hybridized sulfur atoms impart conformational flexibility to optimize Mg2+ coordination. These structural and electronic advantages make 2356PBQS a high-performance cathode material for RMBs.Author contributions
Hongda Gui: writing – original draft, investigation. Ze Wang: investigation. Yuliang Cao: writing – review & editing, software. Fei Xu: writing – review & editing, funding acquisition, conceptualization.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available within the paper and its supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07331f.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (22179101). We thank the Core Facility of Wuhan University for FTIR, XPS and TEM tests. The theoretical calculations in this work have been done on the supercomputing system in the Supercomputing Center of Wuhan University.References
- X. Zhang, X. Sun, X. Li, X. Hu, S. Cai and C. Zheng, J. Energy Chem., 2021, 59, 343–363, DOI:10.1016/j.jechem.2020.11.022.
- J. Deng, C. Bae, A. Denlinger and T. Miller, Joule, 2020, 4, 511–515, DOI:10.1016/j.joule.2020.01.013.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99, DOI:10.1016/j.jechem.2020.10.017.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727, DOI:10.1038/35037553.
- Z. Zhao-Karger, M. E. G. Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820, 10.1039/c7ta02237a.
- Q. Sun, S. Luo, R. Huang, S. Yan and X. Lin, Coord. Chem. Rev., 2024, 515, 215956, DOI:10.1016/j.ccr.2024.215956.
- S. Li, J. Zhang, S. Zhang, Q. Liu, H. Cheng, L. Fan, W. Zhang, X. Wang, Q. Wu and Y. Lu, Nat. Energy, 2024, 9, 285–297, DOI:10.1038/s41560-023-01439-w.
- X. Xue, R. Chen, X. Song, A. Tao, W. Yan, W. Kong and Z. Jin, Adv. Funct. Mater., 2021, 31, 2009394, DOI:10.1002/adfm.202009394.
- A. Roy, M. Sotoudeh, S. Dinda, Y. Tang, C. Kuebel, A. Gross, Z. Zhao-Karger, M. Fichtner and Z. Li, Nat. Commun., 2024, 15, 492, DOI:10.1038/s41467-023-44495-2.
- X. He, R. Cheng, X. Sun, H. Xu, Z. Li, F. Sun, Y. Zhan, J. Zou and R. M. Laine, J. Magnesium Alloys, 2023, 11, 4359–4389, DOI:10.1016/j.jma.2023.11.008.
- X. Ren, D. Tao, Y. Tang, Y. Cao and F. Xu, J. Mater. Chem. A, 2023, 11, 9955–9963, 10.1039/d3ta01293j.
- X. Ren, D. Tao, S. Cui, T. Li, Y. Cao and F. Xu, Energy Storage Mater., 2023, 63, 102992, DOI:10.1016/j.ensm.2023.102992.
- H. Gui, D. Tao, Y. Tang, Y. Cao and F. Xu, ACS Sustain. Chem. Eng., 2024, 12, 17292–17300, DOI:10.1021/acssuschemeng.4c06991.
- Z. Ye, S. Xie, Z. Cao, L. Wang, D. Xu, H. Zhang, J. Matz, P. Dong, H. Fang, J. Shen and M. Ye, Energy Storage Mater., 2021, 37, 378–386, DOI:10.1016/j.ensm.2021.02.022.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, W. F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820, DOI:10.1021/acs.jcim.9b00725.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094, DOI:10.1021/cr00031a013.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041, DOI:10.1063/1.474659.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163, DOI:10.1002/wcms.19.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403, DOI:10.1063/1.458517.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842, DOI:10.1016/S0040-4039(01)94175-9.
- H. L. Schmider and A. D. Becke, J. Mol. Struct.:THEOCHEM, 2000, 527, 51–61, DOI:10.1016/S0166-1280(00)00477-2.
- T. Lu and Q. Chen, Theor. Chem. Acc., 2020, 139, 25, DOI:10.1007/s00214-019-2541-z.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183, DOI:10.1142/S0219633612500113.
- I. Mayer and P. Salvador, Chem. Phys. Lett., 2004, 383, 368–375, DOI:10.1016/j.cplett.2003.11.048.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506, DOI:10.1021/ja100936w.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885.
- T. Lu, J. Chem. Phys., 2024, 161, 82503, DOI:10.1063/5.0216272.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38, DOI:10.1016/0263-7855(96)00018-5.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970.
- Y. Zhao, M. Zhou, X. Zeng, J. Yang, J. Wang and Y. Nuli, Adv. Funct. Mater., 2025, 2423533, DOI:10.1002/adfm.202423533.
- J. Xie, Z. Wang, P. Gu, Y. Zhao, Z. J. Xu and Q. Zhang, Sci. China Mater., 2016, 59, 6–11, DOI:10.1007/s40843-016-0112-3.
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509, DOI:10.1002/aenm.201703509.
- N. Tran, J. Leprêtre and F. Alloin, Electrochim. Acta, 2021, 375, 137990, DOI:10.1016/j.electacta.2021.137990.
- M. Sajid, S. U. Rahman, F. Tao, D. Xie, M. S. U. Rehman, A. Salah and J. Zhang, Batteries Supercaps, 2025, e202400675, DOI:10.1002/batt.202400675.
- G. Dawut, Y. Lu, L. Mao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396, 10.1039/c8qi00197a.
- X. Shi, A. Yi, Q. Liu, Y. Zhang, S. Lin and X. Lu, ACS Nano, 2023, 17, 25005–25013, DOI:10.1021/acsnano.3c07346.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124, DOI:10.1002/advs.201500124.
- Z. Hu, L. Huang, X. Gan, Y. Han, J. Chu and Z. Song, ACS Appl. Mater. Interfaces, 2024, 16, 19014–19025, DOI:10.1021/acsami.4c02399.
- D. Tao, T. Li, Y. Tang, H. Gui, Y. Cao and F. Xu, Adv. Funct. Mater., 2024, 34, 2411223, DOI:10.1002/adfm.202411223.
- Y. Zhao, S. Chen, M. Zhou, M. Pan, Y. Sun, D. Zhang, S. Zhang, Y. Wang, M. Li, X. Zeng, J. Yang, J. Wang and Y. Nuli, ACS Nano, 2024, 18, 22356–22368, DOI:10.1021/acsnano.4c06653.
- H. Dong, Y. Liang, O. Tutusaus, R. Mohtadi, Y. Zhang, F. Hao and Y. Yao, Joule, 2019, 3, 782–793, DOI:10.1016/j.joule.2018.11.022.
- Q. Su, W. Wang, J. Chen, J. Ji, W. Wang, W. Ren, L. Zhang, J. Xie and Q. An, Adv. Funct. Mater., 2024, 35, 2419594, DOI:10.1002/adfm.202419594.
- D. Tao, L. Ran, T. Li, Y. Cao and F. Xu, ACS Nano, 2024, 18, 28810–28821, DOI:10.1021/acsnano.4c08576.
- Z. Li, B. P. Vinayan, P. Jankowski, C. Njel, A. Roy, T. Vegge, J. Maibach, J. M. G. Lastra, M. Fichtner and Z. Zhao-Karger, Angew. Chem., Int. Ed., 2020, 59, 11483–11490, DOI:10.1002/anie.202002560.
- X. Ren, F. Wen, S. Cao and F. Xu, Chem. Eng. J., 2023, 452, 139570, DOI:10.1016/j.cej.2022.139570.
- S. Cui, T. Li, D. Tao, D. Zhang, Y. Cao and F. Xu, ACS Mater. Lett., 2024, 6, 1883–1889, DOI:10.1021/acsmaterialslett.4c00530.
- S. Cui, T. Li, D. Tao, D. Zhang, Y. Cao and F. Xu, Energy Storage Mater., 2024, 71, 103672, DOI:10.1016/j.ensm.2024.103672.
- V. Prabhakaran, G. Agarwal, J. D. Howard, S. Wi, V. Shutthanandan, D. Nguyen, L. Soule, G. E. Johnson, Y. Liu, F. Yang, X. Feng, J. Guo, K. Hankins, L. A. Curtiss, K. T. Mueller, R. S. Assary and V. Murugesan, ACS Appl. Mater. Interfaces, 2023, 15, 7518–7528, DOI:10.1021/acsami.2c18477.
- Y. Mu, J. Nyakuchena, Y. Wang, J. R. Wilkes, T. Luo, M. Goldstein, B. Elander, U. Mohanty, J. L. Bao, J. Huang and D. Wang, Angew. Chem., Int. Ed., 2024, 63, e202409286, DOI:10.1002/anie.202409286.
| This journal is © The Royal Society of Chemistry 2026 |