DOI:
10.1039/D5SC07306E
(Edge Article)
Chem. Sci., 2026,
17, 3122-3128
High resolution photoelectron imaging of cryogenically cooled alkaline-earth metal complexes with the BO2 superhalogen, MBO2− (M = Ca, Sr, Ba)
Received
20th September 2025
, Accepted 12th December 2025
First published on 15th December 2025
Abstract
The BO2 molecule is a superhalogen with a very high electron affinity, resulting in an extremely stable BO2− anion suitable as a building block to form ionic compounds. Here we report the generation of M(BO2−) (M = Ca, Sr, Ba) complexes and the investigation of their structures and bonding using high-resolution cryogenic photoelectron imaging. All three M(BO2−) alkaline-earth complexes are found to have linear M(O–B–O−) (1Σ+) structures. Photodetachment removes an electron from the alkaline-earth metal atom and produces the neutral M+(O–B–O−) (2Σ+) ionically bonded ground state. The change of the charge state on the metal center induces a significant reduction of the M–O bond length in the neutral final state, resulting in an extensive M–O stretching vibrational progression in all the photoelectron spectra. The electron affinities of MBO2 are measured to be 1.574 eV, 1.487 eV, and 1.291 eV and the M–O stretching frequencies are measured to be 411 cm−1, 339 cm−1, and 290 cm−1 for M = Ca, Sr, and Ba, respectively. The strong electron-withdrawing power of BO2 leads to the ionically bonded ground state for MBO2 (2Σ+), resulting in a single electron localized on the metal center. The ionic interaction between M+ and BO2− in MBO2 makes their low-lying electronic excitations resemble atomic transitions, rendering MBO2-type molecules promising candidates for laser cooling.
1 Introduction
The BO2 molecule is electron-deficient. Adding an electron to BO2 gives rise to a very stable BO2− anion, which is isoelectronic with CO2. The electron affinity (EA) of BO2 was measured by photoelectron spectroscopy (PES) to be 4.46 eV,1 making it a member of the superhalogen family.2 The highly stable BO2− anion has been used as a building block to design novel ionic complexes and compounds.3–5 Recently, the alkaline-earth metal complexes with BO2−, M+(BO2−), have attracted interest as promising molecular candidates for laser cooling and ultracold chemistry.6 Laser cooling relies on repeated photon cycling to slow the translational motion of the molecule. Unlike atoms, however, molecules can be lost from the cooling cycle due to vibrational branching. Thus, the key requirement of a laser-coolable molecule is highly diagonal Franck–Condon factors (FCFs).7–9 The high EA of BO2 can take one electron away from the alkaline-earth metal M, leaving an isolated M+ with an unpaired s electron and an M+(BO2−) ionically bonded ground state. Low-lying electronic excitations in MBO2 involve essentially the atomic transitions of the metal cation M+, with the BO2− ligand being a spectator. To maximize diagonal FCFs, the unpaired electron on M+ should be as localized as possible, favouring ligands with strong electron-withdrawing power and little covalent interaction with the metal. Recent theoretical calculations show that, among many commonly considered ligands, BO2 has the highest EA and the strongest electron-withdrawing ability.6 Thus, the MBO2 species exhibit the smallest M–O bond length changes upon electronic excitations – the condition to produce diagonal FCFs. However, alkaline-earth MBO2 complexes have not been studied experimentally and their spectroscopic properties are not known.
On the other hand, there have been extensive studies on the spectroscopy of metal complexes of the isoelectronic CO2 molecule.10,11 These complexes are especially interesting as simple models to understand CO2 activation at the atomic level.12–26 Negatively charged metal ions, such as Cu−, Ag−, Au−, Ni−, Pd−, Pt−, and Bi−, can form complexes with CO2 by attaching to the carbon atom.13–20 In contrast, positively charged metal ions, such as Mg+, Ca+, V+, and Co+, bind with CO2 to form weakly bound, linear M+(O–C–O) complexes.21–26 In addition, cyclic structures, in which metals bind simultaneously to both oxygen atoms, have also been observed for neutral alkali metals with CO2 in low-temperature matrices.27,28 Despite the rich structures that metal-CO2 complexes can form, the chemistry of isoelectronic metal-BO2− complexes was much less explored. Only a few low-resolution PES studies have been reported on MBO2− complexes with M = Cu, Ag, Au, Fe, Al, Na, and Li.3,29–34
Motivated by the potential applications of the alkaline-earth metal MBO2 species in laser cooling and the interest in understanding their structures and bonding, we present a high-resolution photoelectron imaging (PEI) study of MBO2− (M = Ca, Sr, Ba). In our PEI experiment, an electron is detached from the MBO2− anion, yielding spectroscopic information about the MBO2 neutral final states. Our high-resolution PEI apparatus was recently equipped with a cryogenic ion trap to create cold cluster anions produced from a laser vaporization source.35 Cold anions are critical for high-resolution PEI, as demonstrated in recent studies on boron-related clusters.36–40 In the current work, we have obtained vibrationally resolved photoelectron (PE) spectra for all three MBO2− alkaline-earth complexes. Both the anion and the neutral species are shown to be linear. In addition to measuring accurate EAs, we are able to obtain vibrational information for the M–O stretching mode and the B–O stretching modes in the BO2 moiety. The observation of extensive M–O stretching progression in each case confirms electron detachment from the metal to yield an ionically bonded neutral MBO2, i.e., M+(BO2−). Experimental and theoretical evidence is presented to show that the atomic nature of M+ is maintained with BO2− being a spectator, which is a key feature of good molecular candidates for laser cooling.
2 Experimental and theoretical methods
2.1 Photoelectron imaging
The experiment was carried out using a high-resolution PEI apparatus recently equipped with a cryogenically cooled 3D Paul trap.35 Details of the PEI apparatus were described previously.41 In this study, the MBO2− (M = Ca, Sr, Ba) complexes were produced by laser vaporization of cold-pressed M/11B targets, made from the respective metal and 11B-enriched boron powders. Oxygen impurities always existed on the target surface, which could give strong MBO2− mass signals especially for fresh targets. To produce stable and consistent MBO2− complexes, we employed a helium carrier gas seeded with 0.1% O2. Clusters formed inside the nozzle were entrained by the carrier gas and underwent a supersonic expansion. After passing through a skimmer, the collimated cluster beam travelled directly into the downstream 3D Paul trap cooled to 4.2 K using a two-stage closed-cycle helium refrigerator. The buffer gas (helium with 20% H2) was pulsed into the Paul trap on the arrival of the clusters. Negatively charged clusters were trapped and cooled by collisions with the buffer gas. After a 45 ms trapping/cooling time, the cold clusters were extracted into a time-of-flight mass spectrometer. Clusters of interest were mass-selected and intercepted by a detachment laser in the interaction zone of a velocity map imaging system. Photodetachment was performed using the output of a dye laser pumped using a Nd:YAG laser. Photoelectrons were projected onto a microchannel plate coupled with a phosphor screen and the images were recorded using a charge-coupled device camera. The raw images were analysed using the MEVELER method to retrieve the electron kinetic energy distributions.42 A high resolution of 1.2 cm−1 could be achieved for very slow electrons, while for fast electrons, the relative kinetic energy (Ek) resolution (ΔEk/Ek) was approximately 0.6%.41
PEI also yields information about the photoelectron angular distributions (PADs), which are characterized by the anisotropy parameter β. Under the electric dipole approximation, when an electron is ejected from an s atomic orbital with zero angular momentum (l = 0), the outgoing electron is a p wave (l = 1) with β = 2. When an electron is ejected from a p atomic orbital (l = 1), the outgoing electron carries a mixture of s (l = 0) and d (l = 2) partial waves with β = −1.43 While the β value is non-trivial to interpret for electron detachment from molecular orbitals (MOs), it provides qualitative information about the symmetries of the MOs.44
2.2 Theoretical methods
Structure searches were done for the MBO2− species using the ABCluster software.45 For each system, many initial structures were generated and subsequently optimized using density functional theory (DFT) at the PBE0/def2-SVP level46,47 for both singlet and triplet states. The low-lying isomers were further optimized at the PBE0/def2-TZVP46,47 level, as presented in Fig. S1. The EAs were computed using the energy differences between the optimized neutral and the anion of the global minimum (GM) structures at the PBE0/def2-TZVP level. Single-point calculations at the CCSD(T)/def2-TZVP level of theory were also performed on the GM structures and the corresponding neutrals to obtain more accurate EAs. Frequency calculations were carried out to compare with the experiment and to ensure that the optimized structures were true minima. All electronic structure calculations were done using the Gaussian 09 package.48 FCF calculations were performed using the ezFCF package.49
3 Experimental and theoretical results
3.1 PEI and PES of CaBO2−
High-resolution PE images and spectra of CaBO2− are measured at 1.599 eV, 1.727 eV, and 1.905 eV, as shown in Fig. 1. The 1.905 eV spectrum (Fig. 1c) reveals extensive vibrational structures. The series of peaks, labelled as 000, a, b, and d, display similar spacings, which define a vibrational progression. Next to peak d, there is a discernible feature, which is labelled as peak c. Two weak peaks, e and f, are also observed at higher binding energies. The spacing between peaks e and f is similar to the vibrational progression defined by peaks 000, a, b, and d, suggesting that peak e is due to a high frequency mode and peak f is its combinational level. A weak hot band, labelled as hb, is also observed on the lower binding energy side of the 000 peak. The hot band yields a vibrational frequency of 331 cm−1 for the CaBO2− anion. In the 1.727 eV spectrum (Fig. 1b), peaks 000, a, and b are better resolved. In the 1.599 eV spectrum (Fig. 1a), the 000 peak, which corresponds to the transition from the vibrational ground state of the anion to that of the neutral ground state, defines an accurate EA of 1.574 eV for CaBO2. The PADs of all the observed vibrational peaks exhibit distinct p wave characters, implying that the detached electron originates from an s or σ-type orbital. Note that the β values depend on the electron kinetic energies (Table 1). The β value of 1.7 for the 000 transition is very close to the ideal value of 2 for electron detachment from atomic s orbitals, indicating the near atomic-like highest occupied molecular orbital (HOMO) of CaBO2−, from which an electron is removed in the PEI experiment. The binding energies of all the vibrational peaks, their shifts relative to peak 000, their assignments, and β values are given in Table 1.
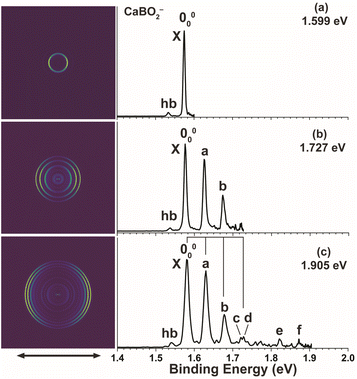 |
| | Fig. 1 Photoelectron images and spectra of CaBO2− at (a) 1.599 eV, (b) 1.727 eV, and (c) 1.905 eV. The double arrow below the images indicates the laser polarization. | |
Table 1 The measured binding energies (BEs), assignments, and shifts relative to the 000 peak for the vibrational features resolved in the spectra of CaBO2−
|
|
BE (eV)a |
Assignment |
Shift (cm−1) |
β
|
|
The numbers in the parentheses represent the uncertainty in the last digit.
The β values are obtained from the 1.905 eV spectrum.
|
| hb |
1.533(3) |
310 |
−331 |
|
| 000 |
1.574(1) |
2Σ+ (000) |
0 |
1.7 |
| a |
1.625(3) |
301 |
411 |
1.6 |
| b |
1.675(1) |
302 |
815 |
1.3 |
| c |
1.722(5) |
201 |
1194 |
|
| d |
1.727(5) |
303 |
1234 |
|
| e |
1.823(3) |
101 |
2008 |
|
| f |
1.872(3) |
101301 |
2404 |
|
3.2 PEI and PES of SrBO2−
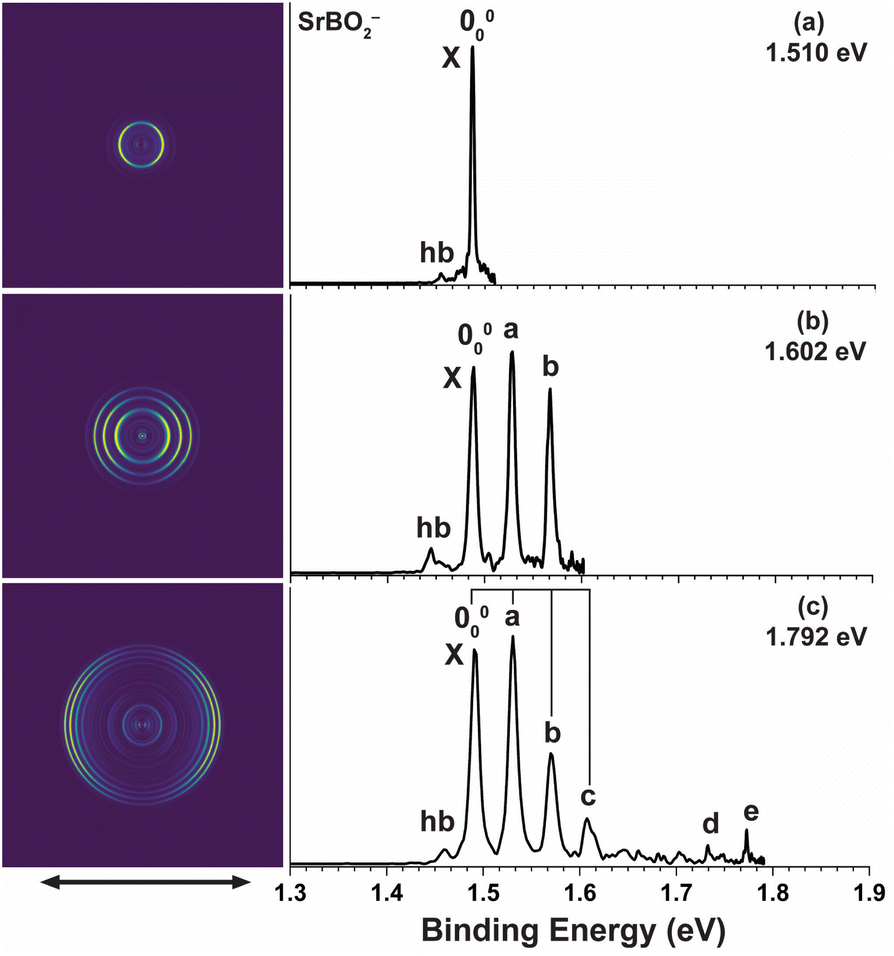
High-resolution PE images and spectra of SrBO2− measured at 1.510 eV, 1.602 eV, and 1.792 eV are presented in Fig. 2. The 1.792 eV spectrum (Fig. 2c) exhibits a vibrational progression closely resembling that for CaBO2−. The main progression is represented by peaks 000, a, b, and c, with similar spacings. Similarly, peak d suggests a high frequency mode and peak e indicates its combinational level. A vibrational hot band (labelled hb) is also observed, giving a vibrational frequency of 258 cm−1 for the anion. The 1.602 eV spectrum (Fig. 2b) resolves peaks 000, a, and b better and the 1.510 eV spectrum (Fig. 2a) defines an accurate EA of 1.487 eV for SrBO2. All observed vibrational peaks exhibit distinct p wave angular distributions with the β values close to 2 for atomic s orbitals, similar to that in CaBO2−, suggesting electron detachment from an s or σ-type orbital. The binding energies of all the observed peaks, their shifts relative to peak 000, their assignments, and β values are given in Table 2.
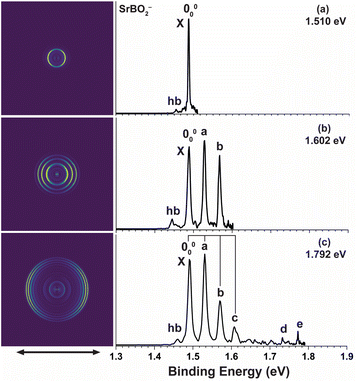 |
| | Fig. 2 Photoelectron images and spectra of SrBO2− at (a) 1.510 eV, (b) 1.602 eV, and (c) 1.792 eV. The double arrow below the images indicates the laser polarization. | |
Table 2 The measured binding energies (BEs), assignments, and shifts relative to the 000 peak for the vibrational features resolved in the spectra of SrBO2−
|
|
BE (eV)a |
Assignment |
Shift (cm−1) |
β
|
|
The numbers in the parentheses represent the uncertainty in the last digit.
The β values are obtained from the 1.792 eV spectrum.
|
| hb |
1.455(3) |
310 |
−258 |
|
| 000 |
1.487(1) |
2Σ+ (000) |
0 |
1.6 |
| a |
1.529(3) |
301 |
339 |
1.6 |
| b |
1.568(1) |
302 |
653 |
1.3 |
| c |
1.611(5) |
303/201 |
1000 |
|
| d |
1.734(3) |
101 |
1992 |
|
| e |
1.774(3) |
101301 |
2315 |
|
3.3 PEI and PES of BaBO2−
High-resolution PE images and spectra of BaBO2− at 1.432 eV and 1.579 eV are presented in Fig. 3. An extensive vibrational progression is revealed, similar to those of CaBO2− and SrBO2−. The main progression is represented by peaks 000, a, b, c, and d with nearly equal spacing. The 000 transition defines an EA of 1.291 eV for BaBO2. The weak hot band (labelled hb) gives an anion vibrational frequency of 242 cm−1. All observed vibrational peaks also exhibit distinct p wave angular distributions with β values close to 2 for s atomic orbitals, similar to those of CaBO2− and SrBO2−, indicating detachment from an s or σ-type orbital. The two PE spectra for BaBO2− in Fig. 3 each show a sharp threshold peak, probably due to threshold enhancement of part of a threshold vibronic transition, possibly involving the sequence bands of the hot band transitions. The binding energies of the observed vibrational peaks, their shifts relative to peak 000, their assignments, and β values are given in Table 3.
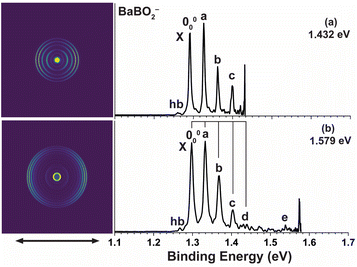 |
| | Fig. 3 Photoelectron images and spectra of BaBO2− at (a) 1.432 eV and (b) 1.579 eV. The double arrow below the images indicates the laser polarization. | |
Table 3 The measured binding energies (BEs), assignments, and shifts relative to the 000 peak for the vibrational features resolved in the spectra of BaBO2−
|
|
BE (eV)a |
Assignment |
Shift (cm−1) |
β
|
|
The numbers in the parentheses represent the uncertainty in the last digit.
The β values are obtained from the 1.579 eV spectrum (Fig. 3b).
|
| hb |
1.261(3) |
310 |
−242 |
|
| 000 |
1.291(3) |
2Σ+ (000) |
0 |
1.6 |
| a |
1.327(3) |
301 |
290 |
1.5 |
| b |
1.362(3) |
302 |
573 |
1.4 |
| c |
1.399(3) |
303 |
871 |
|
| d |
1.436(5) |
304 |
1170 |
|
| e |
1.540(5) |
101 |
2008 |
|
3.4 Theoretical results
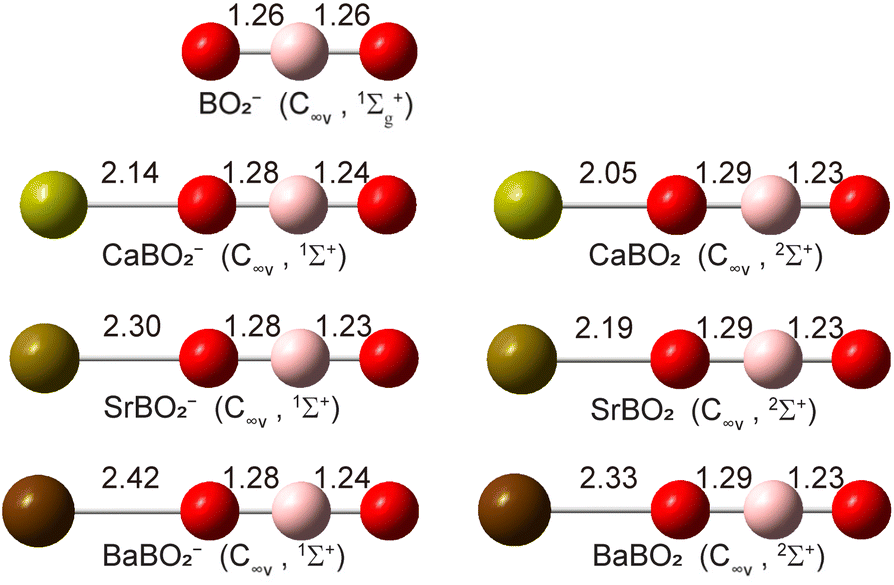
Structure searches (Fig. S1) find that the closed-shell linear M–O–B–O− structure with C∞v symmetry (1Σ+) is overwhelmingly the global minimum for all MBO2− species, as shown in Fig. 4. The B–O bond lengths are all similar to those in bare BO2− (shown in Fig. 4 for comparison): the B–O bond close to the metal is slightly elongated while the B–O bond away from the metal is slightly shortened. The molecular orbitals (MOs) for MBO2− are given in Fig. S2. The HOMO of MBO2− is essentially the valence s orbital on the metal atom, polarized only slightly by the BO2− ligand. Removing an electron from the HOMO gives rise to the doublet 2Σ+ ground state of neutral MBO2 (Fig. 4). The B–O bond lengths remain almost the same in the neutral as in the anion, but the M–O bond length is significantly shortened, which should induce a large vibrational progression in the PE spectra in the M–O stretching mode. The calculated EAs for MBO2 (M = Ca, Sr, Ba), using single-point energies calculated at the CCSD(T)/def2-TZVP level for the linear GM structures, are compared with the corresponding experimental values in Table 4. The calculated vibrational frequencies for the stretching modes of neutral MBO2, scaled by a factor of 0.96,50 are also compared with the experimental data in Table 4.
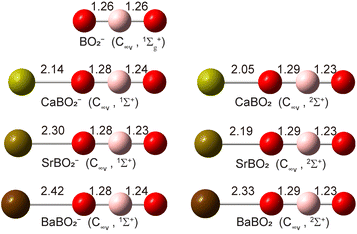 |
| | Fig. 4 The optimized structures of CaBO2−, SrBO2−, BaBO2− and their neutrals at the PBE0/def2-TZVP level. The optimized structure of BO2− is also shown for comparison. The point group symmetries and electronic states are also given. The bond lengths are given in Å. | |
Table 4 The experimental EAs and vibrational frequencies compared with the calculated values for MBO2 (M = Ca, Sr, Ba)
|
|
EA (eV) |
Frequency (cm−1)a |
| Exp. |
Cal.b |
Exp.c |
Cal. |
|
Calculated frequencies at the PBE0/def2-TZVP level are scaled by a factor of 0.96. The vibrational displacement vectors are shown in Fig. S2.
At the CCSD(T)/def2-TZVP level. The EAs computed at the PBE0/def2-TZVP level are 1.48, 1.35, and 1.22 eV for M = Ca, Sr, and Ba, respectively.
The experimental uncertainty is estimated to be ±10 cm−1.
|
| CaBO2 |
1.574 |
1.52 |
411 (ν3) |
372 |
| 1193 (ν2) |
1105 |
| 2008 (ν1) |
1978 |
| SrBO2 |
1.487 |
1.45 |
339 (ν3) |
300 |
| 992 (ν2) |
1093 |
| 1992 (ν1) |
1990 |
| BaBO2 |
1.291 |
1.23 |
290 (ν3) |
275 |
| 2008 (ν1) |
1998 |
4 Discussion
4.1 Comparison between experiment and theory
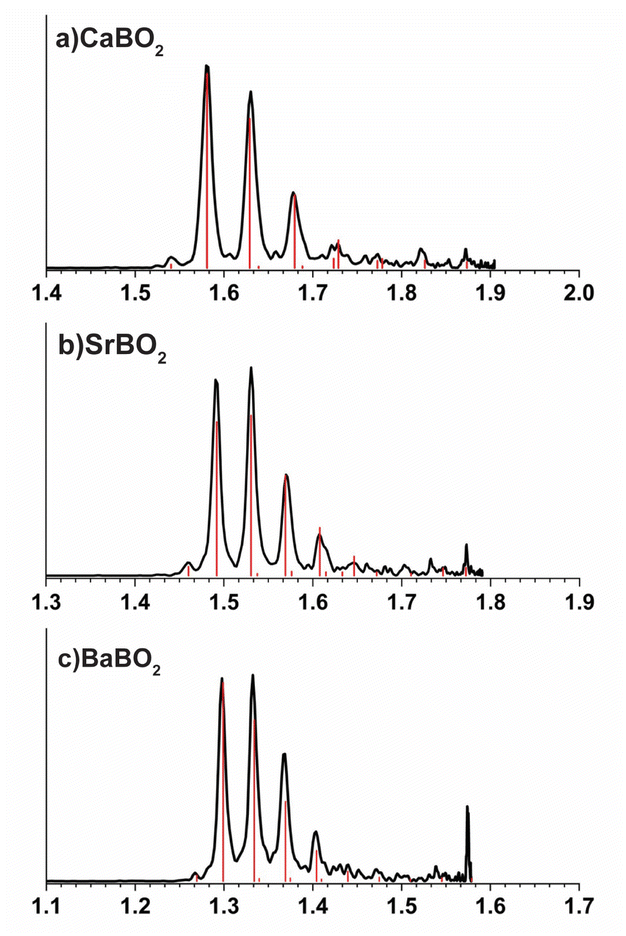
The calculated EAs for the linear GM structures of CaBO2, SrBO2, and BaBO2 at the CCSD(T)/def2-TZVP level are 1.52 eV, 1.45 eV, and 1.23 eV, and they are in excellent agreement with the experimental data of 1.547 eV, 1.487 eV and 1.291 eV, respectively, as shown in Table 4. To understand the observed vibrational structures, we computed the FCFs using the optimized geometries and vibrational frequencies of the anions and neutrals at the PBE0/def2-TZVP level, as shown in Fig. 5. The most FC-active mode is the M–O stretching (ν3 in Fig. S3), as expected. The calculated FCFs are in excellent agreement with the PE spectra, allowing all the vibrational features to be readily assigned.
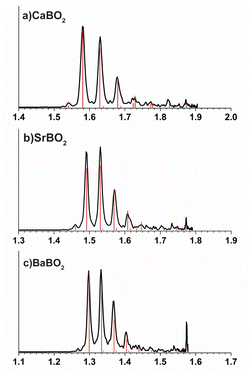 |
| | Fig. 5 Calculated Franck–Condon factors for the ground state detachment transitions of (a) CaBO2−, (b) SrBO2−, and (c) BaBO2−. | |
The extensive ν3 vibrational progression in all three systems agrees with the large change in the M–O bond length upon electron detachment. The observed hot band in each spectrum comes from the FC-active M–O stretching mode of the anions, measured to be 331 cm−1, 258 cm−1, and 242 cm−1 for CaBO2−, SrBO2−, and BaBO2−, respectively. The M–O stretching frequencies in the anions are all much smaller than those in the neutrals (Table 4), as expected. The FCF calculations suggest a vibrational temperature of ∼100 K, which is higher than we expected, probably due to gas condensation in the interior of the ion trap. The presence of the weak hot band, nonetheless, did not affect the resolution of the PE spectra, but allowed the anion vibrational frequencies to be measured.
The computed FCFs suggest that peak c in the CaBO2− spectrum can be assigned to the ν2 mode, that is, the symmetric stretching of the O–B–O moiety (Fig. S3); peak e is due to the ν1 mode, which is the asymmetric stretching of the O–B–O moiety (Fig. S3), while peak f is the combinational level between ν1 and ν3. Similarly, in the SrBO2− spectrum, peak d can be assigned to the ν1 mode and peak e is due to the combinational level between ν1 and ν3. In the BaBO2− spectrum, peak e can be assigned to the ν1 mode. There are only three linear stretching modes for the linear M–O–B–O molecules. We are able to observe all of them for CaBO2. The weak FC activities for the ν1 and ν2 modes are due to the very small B–O bond length changes from the anion to the neutral (Fig. 4). Note that the asymmetric O–B–O stretching frequency and symmetric O–B–O stretching frequency in bare BO2− are 1992 cm−1 and 1110 cm−1, respectively, which are similar to those observed in CaBO2. For SrBO2, we are only able to resolve the ν1 transition; the ν2 vibration is very close to the 303 transition (peak c, Fig. 2c). In the case of BaBO2, the ν2 vibrational frequency is very close to the 304 transition (peak d, Fig. 3b). Overall, the excellent agreement of the calculated EAs and the calculated FCFs with the experimental data unequivocally confirms the linear structures for both the MBO2− anion and MBO2 neutral species.
4.2 Chemical bond analyses
The computed structures (Fig. 4) and the experimental vibrational information all indicate that the BO2− anion moiety remains intact in both the MBO2− anion and the MBO2 neutral. In addition to the atomic-like σ HOMO, we find that the remaining valence MOs of the BO2− moiety in MBO2− are similar to those of the bare BO2− anion, as shown in Fig. S2. Natural charge analyses were carried out for CaBO2−, SrBO2−, and BaBO2− and their neutrals, as shown in Fig. 6. In the MBO2− anions, the metal atom is essentially neutral, carrying negligible charges, whereas the BO2 moiety retains the unit negative charge. The intact nature of BO2− is further supported by the similarity of the B–O bond lengths in MBO2− and MBO2 compared with those in bare BO2− (Fig. 4).
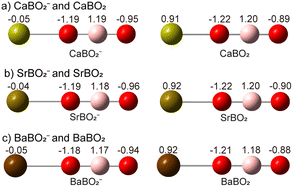 |
| | Fig. 6 Natural charge distributions for (a) CaBO2− and CaBO2, (b) SrBO2− and SrBO2, and (c) BaBO2− and BaBO2. | |
Thus, the MBO2− anion is essentially a neutral alkaline-earth atom interacting weakly with the BO2− anion. The negative charge on the ligand polarizes the spherically symmetric valence s electron cloud, inducing a dipole moment on the metal and giving rise to a dipole–charge interaction. This polarization interaction distorts the charge distribution on BO2− with more negative charge pulled to the oxygen atom bound to the metal. The induced-dipole–charge interaction is the reason why the linear M–O–B–O− geometry is favoured.
Photodetachment from the HOMO of the MBO2− anion, which is essentially the metal valence s orbital, leads to the 2Σ+ neutral ground state, leaving one s electron localized on the alkaline-earth metal cation. Natural charge analyses indicate that, in the neutral MBO2 complexes, the metal carries nearly a unit positive charge (Fig. 6 and Table S1), while the BO2− ligand retains its unit negative charge. Thus, neutral MBO2 is a completely ionic molecule between M+ and BO2−, leading to the significantly shortened M–O bond length compared to that in the anion due to the strong Coulombic interaction. This large bond-length contraction gives rise to the pronounced M–O stretching progressions in the PE spectra. We analysed the spin densities of MBO2, showing that the unpaired spin is nearly completely localized on the metal (Table S1). The ionic bonding nature between M+ and BO2− in MBO2 is further supported by the Wiberg bond indices (Table S2), which indicate negligible covalent bonding between the M+ and BO2− fragments. It is interesting to note that ionization of the valence s electron from M in M(BO2−) requires much less energy in comparison to the ionization energies of the bare metal atoms (6.11 eV for Ca, 5.69 eV for Sr, and 5.21 eV for Ba) due to the Coulombic repulsion from the negatively charged BO2− ligand.
4.3 Implications for laser cooling
The alkaline-earth MBO2 complexes are promising laser cooling candidates because of the superhalogen property of BO2 that results in an ionic M+(BO2−) species and leaves a single isolated s electron on the alkaline-earth metal cation M+. Thus, the cycling transition happens on the metal centre with highly diagonal FCFs while the BO2− ligand is a spectator. Note that the current PEI experiments do not directly probe the MBO2 excited states within the cycling transition; the extensive FCFs observed in the current study correspond to photodetachment transitions from the MBO2− anion to neutral MBO2. Nevertheless, the PEI experiment provides valuable information about the structures and vibrational frequencies of the MBO2 neutral ground state and the nature of the atomic-like HOMO on the metal site. The observed large M–O stretching progression confirms the cationic nature of M+ and its dominating ionic interaction with BO2−. The angular distributions of the PE images and the β values show that the photodetachment transition comes from a nearly ideal s orbital on the metal site, because of the electrostatic nature of the metal–ligand interactions in both MBO2− and MBO2. It is expected that the electrostatic interaction in M+(BO2−) would not be affected significantly upon the s to p cycling transition, making the MBO2 systems excellent laser cooling targets. Indeed, recent theoretical calculations predict that the alkaline-earth MBO2 complexes exhibit very small M–O bond length changes upon s to p excitation,6 conducive to yielding highly diagonal FCFs. Additionally, the current study shows that the MBO2 complexes can be readily generated in a laser vaporization cluster source. The number density of the neutral MBO2 species from the laser vaporization source is expected to be much higher than that of the MBO2− anions used for the current PEI experiments.
5 Conclusion
In conclusion, we report an investigation of CaBO2−, SrBO2− and BaBO2− and their corresponding neutrals (MBO2) using high-resolution photoelectron imaging. The electron affinities of neutral CaBO2, SrBO2, and BaBO2 are measured to be 1.574 eV, 1.487 eV and 1.291 eV, respectively. Vibrationally resolved photoelectron imaging, combined with computational chemistry, confirms that the MBO2− anions have linear M–O–B–O− structures with a singlet 1Σ+ ground state and the MBO2 neutrals all have ionically bonded linear M+[O–B–O]− structures with a doublet (2Σ+) ground state. Chemical bond analyses show that the MBO2− anion consists of essentially a neutral alkaline-earth atom weakly interacting with a BO2− ligand. Photodetachment removes an electron from the metal atom, resulting in the M+(BO2−) ionic compound for neutral MBO2. The M–O stretching vibrational frequencies for all three MBO2 complexes are measured, and in the case of CaBO2, both the B–O symmetric and asymmetric stretching frequencies are also measured. The atomic nature of the HOMO in MBO2 is also revealed from the angular distributions of the photoelectron images. The current study confirms the recent theoretical study that suggests that alkaline-earth MBO2 complexes are ideal molecular systems for laser cooling.
Author contributions
J. H. led the experiment. H. W. G. and X. Y. Z. helped with the experiment. J. H. did the calculations. H. W. G. and J. H. analysed the data. H. W. G. led the writing of the manuscript. L. S. W. guided and advised the project and revised and finalized the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data that supports the findings of this study is available from the corresponding author upon request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc07306e.
Acknowledgements
This work was supported by the U.S. National Science Foundation (Grant No. CHE-2403841). The calculations were performed using resources at the Centre for Computation and Visualization (CCV) of Brown University.
References
- H. J. Zhai, L. M. Wang, S. D. Li and L. S. Wang, J. Phys. Chem. A, 2007, 111, 1030 CrossRef CAS PubMed.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277 CrossRef CAS.
- M. Willis, M. Götz, A. K. Kandalam, G. F. Ganteför and P. Jena, Angew. Chem., Int. Ed., 2010, 49, 8966 CrossRef CAS PubMed.
- C. Huang, M. Mutailipu, F. Zhang, K. J. Griffith, C. Hu, Z. Yang, J. M. Griffin, K. R. Poeppelmeier and S. Pan, Nat. Commun., 2021, 12, 2597 Search PubMed.
- S. Liu, X. Jiang, L. Qi, Y. Hu, K. Duanmu, C. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202403328 CrossRef CAS PubMed.
- M. V. Ivanov, F. H. Bangerter and A. I. Krylov, Phys. Chem. Chem. Phys., 2019, 21, 19447 Search PubMed.
- M. D. Rosa, Eur. Phys. J. D, 2004, 31, 395 CrossRef CAS.
- T. A. Isaev and R. Berger, Phys. Rev. Lett., 2016, 116, 063006 CrossRef PubMed.
- S. Vadachkoria, Q. Lei, T. C. Steimle and M. C. Heaven, J. Phys. Chem. Lett., 2025, 16, 3309 CrossRef CAS.
- M. A. Duncan, Int. Rev. Phys. Chem., 2003, 22, 407 Search PubMed.
- N. C. Polfer and J. Oomens, Mass Spectrom. Rev., 2009, 28, 468 CrossRef CAS PubMed.
- D. K. Böhme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44, 2336 CrossRef.
- A. D. Boese, H. Schneider, A. N. Glöß and J. M. Weber, J. Chem. Phys., 2005, 122, 154301 CrossRef.
- B. J. Knurr and J. M. Weber, J. Am. Chem. Soc., 2012, 134, 18804 CrossRef CAS PubMed.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2013, 117, 10764 CrossRef CAS.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2014, 118, 10246 CrossRef CAS.
- X. Zhang, E. Lim, S. K. Kim and K. H. Bowen, J. Chem. Phys., 2015, 143, 174305 CrossRef PubMed.
- M. C. Thompson, J. Ramsay and J. M. Weber, Angew. Chem., Int. Ed., 2016, 55, 15171 CrossRef CAS.
- G. Liu, S. M. Ciborowski, Z. Zhu, Y. Chen, X. Zhang and K. H. Bowen, Phys. Chem. Chem. Phys., 2019, 21, 10955 RSC.
- R. Wang, G. Liu, S. K. Kim, K. H. Bowen and X. Zhang, J. Energy Chem., 2021, 63, 130 CrossRef CAS.
- M. Sodupe, C. W. Bauschlicher and H. Partridge, Chem. Phys. Lett., 1992, 192, 185 Search PubMed.
- C. S. Yeh, K. F. Willey, D. L. Robbins, J. S. Pilgrim and M. A. Duncan, J. Chem. Phys., 1993, 98, 1867 Search PubMed.
- C. T. Scurlock, S. H. Pullins and M. A. Duncan, J. Chem. Phys., 1996, 105, 3579 CrossRef CAS.
- G. Gregoire, J. Velasquez and M. A. Duncan, Chem. Phys. Lett., 2001, 349, 451 CrossRef CAS.
- N. R. Walker, G. A. Grieves, R. S. Walters and M. A. Duncan, Chem. Phys. Lett., 2003, 380, 230 CrossRef CAS.
- N. R. Walker, R. S. Walters and M. A. Duncan, J. Chem. Phys., 2004, 120, 10037 CrossRef CAS.
- Z. H. Kafafi, R. H. Hauge, W. E. Billups and J. L. Margrave, Inorg. Chem., 1984, 23, 177 Search PubMed.
- V. N. Solov’ev, E. V. Polikarpov, A. V. Nemukhin and G. B. Sergeev, J. Phys. Chem. A, 1999, 103, 6721 CrossRef.
- Y. Feng, H. G. Xu, Z. G. Zhang, Z. Gao and W. Zheng, J. Chem. Phys., 2010, 132, 074308 CrossRef PubMed.
- Y. Feng, H. G. Xu, W. Zheng, H. Zhao, A. K. Kandalam and P. Jena, J. Chem. Phys., 2011, 134, 094309 CrossRef.
- X. Y. Kong, H. G. Xu, P. Koirala, W. J. Zheng, A. K. Kandalam and P. Jena, Phys. Chem. Chem. Phys., 2014, 16, 26067 RSC.
- G. L. Gutsev, C. A. Weatherford, L. E. Johnson and P. Jena, J. Comput. Chem., 2012, 33, 416 CrossRef CAS.
- Y. Feng, M. Cheng, X. Y. Kong, H. G. Xu and W. J. Zheng, Phys. Chem. Chem. Phys., 2011, 13, 15865 RSC.
- Z. Zeng, G. L. Hou, J. Song, G. Feng, H. G. Xu and W. J. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 9135 RSC.
- G. S. Kocheril, H. W. Gao, D. F. Yuan and L. S. Wang, J. Chem. Phys., 2022, 157, 171101 CrossRef CAS PubMed.
- H. W. Gao, H. W. Choi, J. Hui, W. J. Chen, G. S. Kocheril and L. S. Wang, J. Chem. Phys., 2023, 159, 114301 Search PubMed.
- H. W. Gao, H. W. Choi, J. Hui and L. S. Wang, J. Phys. Chem. A, 2024, 128, 3579 CrossRef CAS.
- H. W. Gao, J. Hui and L. S. Wang, Chem. Commun., 2023, 59, 12431 RSC.
- H. W. Gao, J. Hui and L. S. Wang, J. Phys. Chem. Lett., 2025, 16, 2039 CrossRef CAS.
- H. W. Gao, J. Hui and L. S. Wang, Chem. Sci., 2025, 16, 7004 RSC.
- I. León, Z. Yang, H. T. Liu and L. S. Wang, Rev. Sci. Instrum., 2014, 85, 083106 CrossRef.
- B. Dick, Phys. Chem. Chem. Phys., 2019, 21, 19499 RSC.
- J. Cooper and R. N. Zare, J. Chem. Phys., 1968, 48, 942 CrossRef CAS.
- A. Sanov and R. Mabbs, Int. Rev. Phys. Chem., 2008, 27, 53 Search PubMed.
- J. Zhang and M. Dolg, Phys. Chem. Chem. Phys., 2015, 17, 24173 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 Search PubMed.
-
M. J. Frisch
et al.
, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Gozem and A. I. Krylov, WIREs Comp. Mol. Sci., 2021, 12, e1546 CrossRef.
-
NIST computational chemistry comparison and benchmark database, https://cccbdb.nist.gov/20vibscalejustx.asp.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Jie
Hui†
,
Jie
Hui†