
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative borane activation by tuning hemilability of different Os-κ2-N,Se-chelated complexes
Faneesha
Assanar
,
Sourav
Gayen
,
Deepak Kumar
Patel
,
Thalappil
Pradeep
 * and
Sundargopal
Ghosh
*
* and
Sundargopal
Ghosh
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in; pradeep@iitm.ac.in
First published on 15th December 2025
Abstract
In an effort to investigate the cooperative activation of boranes, we have designed Os(III) based paramagnetic mono- and bis-κ2-1,3-N,Se-chelated complexes, [Os(PPh3)2(κ2-N,Se-L)Cl2], 1 and [Os(PPh3)(κ2-N,Se-L)2(L′)], 2–3 (L = C5H4NSe; 2: L′ = Cl and 3: L′ = κ1-Se-L). These complexes were synthesised by the thermolysis of [Os(PPh3)3Cl2] with the potassium salt of 2-selenopyridine, which also afforded a diamagnetic species, [Os(PPh3)2(κ2-N,Se-L)2], 4. A comprehensive study utilizing EPR spectroscopy, DFT calculations, and both photochemical and electrochemical analyses has elucidated the distinctive electronic characteristics of these complexes. To probe the electronic influence on hemilability, the OsNCSe osmaheterocycle-based paramagnetic complexes featuring a hard Os(III) centre and a soft Se donor were studied for B–H activation, highlighting synergistic hemilability and metal–ligand cooperativity. Treatment of 2 with [BH3·SMe2] yielded the Os(dihydridoborate)(octatrihydridoborate) complex, [Os(PPh3)(κ3-H,H,N-BH3L)(κ2-H,Se-B3H7L)] (5) via ring opening of both Os–Se and Os–N bonds and the Os–borallyl complex, [(PPh3)(H)2Os(η5-B3H5L2)] (6) through cleavage of the Os–N bonds. To further assess steric effects on cooperative borane activation, 2 was treated with the bulky aryl-substituted borane BH2R (R = (CF3)2C6H3), which afforded [(PPh3)Os{κ3-H,Se,Se′-(NHRBSeBHRN)(SeC5H4)2}], 7via dual Os–N bond cleavage and insertion of two BHR units followed by B–Se bond formation. Interestingly, we have isolated a unique vertex-fused metallaborane cluster, 8 from the reaction of 6 with an excess of borane. In stark contrast, the diamagnetic bis-κ2-1,3-N,Se-chelated complex [Os(PPh3)2(κ2-N,Se-L)2], 4 exhibited no tendency to activate sterically demanding boranes. However, using an electronically distinct ligand framework in [Os(PPh3)2(κ2-N,S-C7H4NS2)2] (4mbz), we observed notable B–H activation patterns of BHR2 that led to the isolation of the Os(σ-borate) complex, [Os(PPh3)2(κ2-N,S-C7H4NS2)(κ3-H,S,S′-HBR2(C7H4NS2))] (11), where the steric hindrance from the aryl substituents prevented dual-site activation. The bonding insights of these activated species were investigated on the basis of DFT calculations.
Introduction
Metal–ligand cooperativity (MLC) has experienced immense growth over the past few decades as a powerful mechanistic strategy in the field of bond activation and catalysis. It serves as an alternative to conventional catalysis due to the synergistic interaction between the metal and ligand, which plays a crucial role in facilitating both bond breaking and bond formation processes.1–3 In particular, MLC has been widely investigated in the activation of various E–H (E = H, B, C, N, O or Si) bonds4–10 and has subsequently been employed in a range of catalytic reactions.11–13 In the context of MLC, several strategies have been developed to promote the E–H bond activation process: (a) utilizing a combination of a Lewis acidic metal and a Lewis basic ligand that cooperatively cleave the E–H bond in a synergistic fashion;4,5 (b) employing the inverse arrangement, where Lewis acidic ligands abstract electrons from the donor site of the substrate, with the metal acting as a Lewis base to cooperatively facilitate bond activation;6,7 (c) using complexes containing aromatic chelating ligand frameworks, which can undergo dearomatization through deprotonation to provide a reactive site for cooperative bond activation;8,9 (d) incorporating redox non-innocent ligands10 that either directly mediate the bond activation process or act as electron reservoirs within the catalytic cycle.In the context of type (a), the predominant class of Lewis basic ligands are typically found in a multidentate framework. Notably, metal complexes featuring hemilabile 1,3-chelating homo and hetero bidentate ligands have garnered significant interest in small molecule activation, as a key attribute of these ligands is their ability to provide coordinative unsaturation at the metal centre. For instance, Yamaguchi proposed an Ir(III) complex (I),14 featuring a κ2-N,O-pyridionate ligand as an intermediate species in the oxidant-free catalytic oxidation of alcohols.14a Subsequently, Rauchfuss demonstrated that complex I functioned as a more efficient catalyst in the same context.14b Schafer and Love developed group-9 phosphoramidate complexes with different ancillary ligands (II and III, Chart 1), which promote B–H and C–H activation processes.15 These complexes were subsequently utilized in catalytic chemoselective hydroboration of aldehydes and anti-Markovnikov O-phosphoramidation of alkynes. Interestingly, a proton responsive bifunctional ruthenium complex, IV promoted the facile E–H bond activation in H2 and HBPin across the Ru–O bond.16 Beyond traditional hemilabile ligands, Gessner and co-workers designed a ruthenium-based 1,3-chelated complex (V) with a carbene ligating centre, where substrate activation induces a transition between a carbene and an alkyl ligand without complete cleavage of the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.5a
C bond.5a
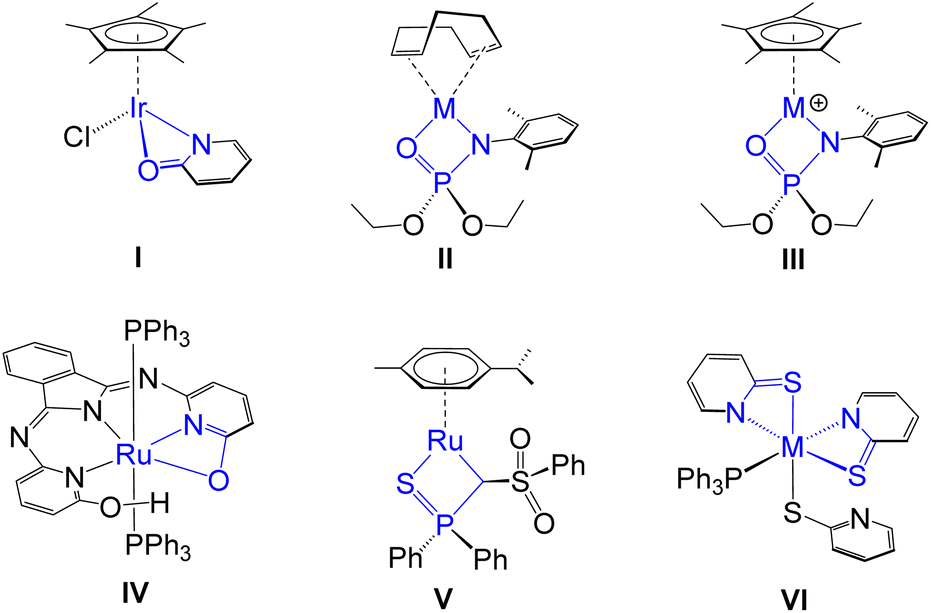 | ||
| Chart 1 Various TM complexes featuring 1,3-chelated hemilabile ligands (M = Rh and Ir for II and III; M = Ru and Os for VI). | ||
In the course of our recent studies, we have synthesized a group of early- and late-transition metal-based mono- and bis-κ2-1,3-N,S-chelated complexes and investigated their role in the cooperative activation of E–H (E = B, C or Si) bonds in small molecules.17–19 The hemilability of four-membered metalla-heterocycles, arising from the coexistence of hard (N) and soft (S) donor sites within polydentate ligands, was pivotal in facilitating E–H bond activation. For instance, we have developed Ru- and Os-based bis-κ2-1,3-N,S-chelated species featuring non-innocent ligands (VI, Chart 1), which upon activation of the simplest borane, [BH3·SMe2], resulted in the formation of unique σ-borate complexes.17b,c However, in the field of MLC, the influence of concerted modulation of electronic properties in both metal and ligand scaffolds remains uncharted. In pursuit of this objective, we have designed different osmaheterocycles by fine-tuning the hardness of both the metal and ligand components. In this study, we synthesised a series of κ2-N,Se-chelated Os(III) complexes such as [Os(PPh3)2(κ2-N,Se-C5H4NSe)Cl2] (1), [Os(PPh3)(κ2-N,Se-C5H4NSe)2Cl] (2) and [Os(PPh3)(κ2-N,Se-C5H4NSe)2(κ1-Se-C5H4NSe)] (3), and engaged them in the cooperative activation of boranes. The perturbed hemilability of the OsNCSe heterocycle led to the cleavage of the Os–N and Os–Se bonds, which facilitated the cooperative activation of free borane. Furthermore, a comparative study was conducted to evaluate the impact of steric and electronic factors by employing both substituted and unsubstituted hydroborane reagents in the cooperative activation process. The activation of sterically encumbered boranes enabled the successful isolation of unique Os–(σ-borate) complexes, utilizing a series of both paramagnetic and diamagnetic Os–N,E-chelated complexes (E = S or Se).
Results and discussion
Recently, we successfully synthesised Cp* based κ2-N,Se-chelated group-6 complexes from the reaction of [Cp*M(CO)3Cl] (M = Mo, W) and [K+L−] (L = C5H4NSe).19 With the objective of synthesizing chelated osmium complexes containing a heavier chalcogen atom, we performed the thermolysis of [Os(PPh3)3Cl2], with excess [KL] at 60 °C. The reaction yielded complexes 1, 2, 3 and 4 which were further isolated in their purest forms by chromatographic techniques (Scheme 1). The complexes were characterized by different spectroscopic methods, mass spectrometry and X-ray diffraction studies. The 1H NMR spectra for complexes 1, 2, and 3 showed several broad resonance peaks in the upfield region, while the 31P{1H} NMR spectra did not reveal any noteworthy signals. In contrast, the 1H NMR spectrum of 4 displayed aromatic signals within the chemical shift range of δ = 8.00–6.25 ppm. Furthermore, species 4 exhibited a single resonance peak at δ = −6.3 ppm in the 31P{1H} NMR spectrum. This detailed analysis suggested that complexes 1, 2, and 3 exhibit paramagnetic behavior, whereas complex 4 appears to be diamagnetic in nature. Mass spectrometric analysis of 1, 2, 3, and 4 indicated that their respective molecular formulae are [Os(PPh3)2(L)Cl2], [Os(PPh3)(L)2Cl], [Os(PPh3)(L)3], and [Os(PPh3)2(L)2]. However, a clear assignment could not be done until the single-crystal X-ray diffraction analysis of these species was carried out.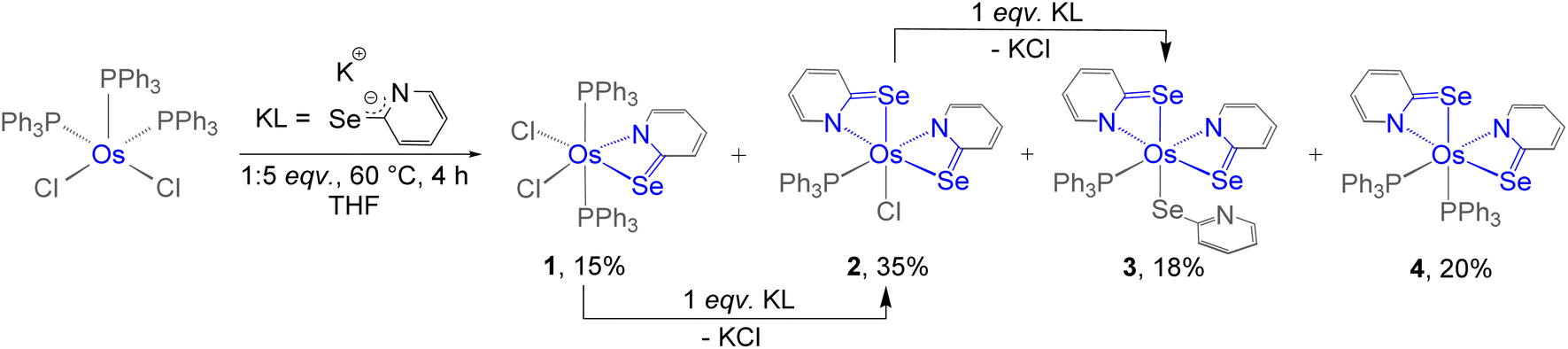 | ||
| Scheme 1 Syntheses of Os(II) and Os(III)–N,Se-chelated complexes, 1–4. | ||
As illustrated in Fig. 1 (left), the molecular structure of complex 1 can be classified as a mono-κ2-N,Se-chelated complex of Os(III), represented as [Os(PPh3)2(κ2-N,Se-L)Cl2]. The molecule exhibited approximately Cs symmetry, with the σ-plane containing the bidentate ligand and two chlorine atoms. On the other hand, the solid-state structure of complex 2 was identified as a bis-κ2-N,Se-chelated Os(III) complex, where the osmium centre is coordinated to two bidentate selenopyridinyl ligands, one chloride, and one PPh3 ligand (Fig. 1, middle). The Os–Cl bond lengths and the bite angles of the osmaheterocycles in complexes 1 and 2 are comparable. The mono-κ2-N,Se-chelated complex 1 was formed through the release of one PPh3 ligand from [Os(PPh3)3Cl2], followed by the coordination of a selenopyridinyl ligand (Scheme 1). The subsequent dissociation of another PPh3 ligand from complex 1 and the binding of an additional unit of ligand (L) with concomitant elimination of KCl afforded complex 2. Although we were unable to obtain any suitable crystals for the XRD analysis of 3, it can be identified as [Os(PPh3)(κ2-N,Se-L)2(κ1-Se-L)] based on mass spectrometry and preliminary spectroscopic data.17c The primary distinction between complexes 2 and 3 stems from the substitution of the chloride ligand with a non-innocent ligand. The formation of complex 3 can be rationalized by the monodentate coordination of the selenopyridinyl ligand subsequent to the elimination of KCl. The X-ray diffraction analysis of a suitable orange crystal of complex 4 revealed an octahedral geometry around the Os centre, which includes two κ2-1,3-N,Se-chelated rings and two triphenylphosphine ligands (Fig. 1, right). This complex was formed by the dissociation of one PPh3 ligand from [Os(PPh3)3Cl2], followed by coordination of two selenopyridinyl ligands with concurrent KCl elimination.
 | ||
| Fig. 1 Molecular structure and labelling diagram of 1 (left), 2 (middle) and 4 (right). Selected bond lengths (Å) and bond angles (°) of 1: Os1–N1 2.093(4), Os1–Se1 2.5518(6), Os1–Cl1 2.3672(13), Os1–Cl2 2.3840(13), Se1–Os1–N1 68.31(13); 2: Os1–N1 2.110(3), Os1–N2 2.098(2), Os1–Se1 2.4510(3), Os1–Se2 2.5351(4), Os1–Cl1 2.3676(8), Se1–Os1–N1 70.16(7), Se2–Os1–N2 68.56(7); 4: Os1–N1 2.154(8), Os1–Se1 2.5494(11), Se1–Os1–N1 68.3(2), and Se2–Os1–N2 68.1(2) (aromatic hydrogens are not shown for better clarity). | ||
To verify the monophasic nature of complexes 1 and 2, powder X-ray diffraction (PXRD) patterns were compared with those simulated from their single-crystal X-ray diffraction (SCXRD) data. The powder XRD patterns of both complexes showed reasonable agreement with the simulated patterns based on their single-crystal structures, confirming the homogeneity of the bulk samples (Fig. S6 and S9). However, a few unidentified low-intensity reflections were observed in both cases, which do not significantly affect the overall bulk phase purity. These results support that the synthesized complexes are largely monophasic and homogeneous.
The paramagnetic properties of complexes 1, 2 and 3 were examined using electron paramagnetic resonance (EPR) spectroscopy at both room temperature and low temperature. While the room temperature EPR spectra of these species in CH2Cl2 (1 and 2) and toluene (3) solution were inconclusive, the X-band frozen glass EPR spectra provided valuable insights. As shown in Fig. 2(a) and (c), the EPR spectra of complexes 1 and 2 exhibited a rhombic pattern with three distinct g values. For both complexes, the gz values (1: 2.724; 2: 2.851) and gy values (1: 2.227; 2: 2.052) were clearly resolved, whereas the gx signal appeared broadened and was observed over a range of 1.2–1.7. The line broadening and reduced spectral resolution of these EPR signals are attributed to the pronounced spin–orbit coupling observed in heavy transition metals such as osmium. Furthermore, density functional theory (DFT) calculations revealed that the Mulliken spin density of the unpaired electron in 1 and 2 is predominantly localized on the Os atom (1: +0.87; 2: +0.71) (Fig. 2(b) and (d)). Notably, a substantial spin density was also detected on the Cl atom in 2, which suggests that coupling with the chloride ligand may have contributed to the observed broadening of the EPR signal. To complement these findings, an Evans NMR study was carried out to determine the solution-state magnetic susceptibility. The experimentally determined magnetic moments (µexp) of 1 (1.68 µB) and 2 (1.80 µB) are in good agreement with the theoretically expected value (1.73 µB). This indicated that both complexes exhibit a low-spin Os(III) (5d5) electronic configuration, consistent with previously reported Os(III) systems.20 Furthermore, the EPR spectrum of 3 exhibited a rhombic pattern with a relatively lower gz value (2.324) compared to 1 and 2 (Fig. 2(e)). In this context, spin density analysis revealed a significant accumulation of spin density on the Se atom of κ2-Se-L, accompanied by a decrease in spin density on the metal atom (Fig. 2(f)). Thus, complex 3 can be described as a superposition of two resonating forms: [(L−)OsIII] and [(L˙)OsII], similar to [M(PPh3)(κ2-N,S-C5H4NS)2(κ1-S-C5H4NS)] (M = Ru, Os).17b,c Due to the pronounced spin–orbit coupling in predominantly metal-centred spin systems like 1 and 2, a higher degree of anisotropy was observed in comparison to 3.
 | ||
| Fig. 2 EPR spectra of 1 (a), 2 (c) and 3 (e); spin density plots of 1 (b), 2 (d) and 3 (f). | ||
Owing to the distinct ligand environments around the Os(III) centre in these paramagnetic complexes, we were interested in exploring their redox properties. Redox potentials were measured by cyclic voltammetry, with all values referenced to the Fc+/Fc couple, as summarized in Table S1. The cyclic voltammograms of complexes 1 and 2 revealed two quasi-reversible redox processes each (1: E1/2 = −0.727 V and 0.737 V; 2: E1/2 = −0.850 V and 0.621 V), well-separated within the electrochemical window (Fig. 3(a) and (b)). The cathodic and anodic waves are attributed to the OsIII/OsII reduction and OsIII/OsIV oxidation processes, respectively. Similar redox behaviour and assignments have been reported for other Os(III) complexes.21,22 Furthermore, the cyclic voltammogram of complex 3 yielded three quasi-reversible redox processes where signals at E1/2 = −0.948 V and 1.123 V correspond to the OsIII/OsII and OsIV/OsIII couples, respectively (Table S1). In addition to this, another quasi-reversible wave at 0.148 V is associated with a redox event centred on the dangling heterocyclic ligand, assigned to the OsII(L˙)/OsIII(L−) redox couple. This behaviour closely resembles that observed in redox non-innocent ligand-based Ru and Os complexes, [M(PPh3)(κ2-N,S-L′)2(κ1-S-L′)] (M = Ru, Os; L′ = C5H4NS, C7H4NS2) which was recently reported by our group.17b,c Furthermore, the cyclic voltammogram of the diamagnetic κ2-N,Se chelated complex, 4 exhibited characteristic redox events associated with the Os(III)/Os(II) and Os(IV)/Os(III) couples, which is consistent with the electrochemical behaviour observed for its sulfur analogue.17c
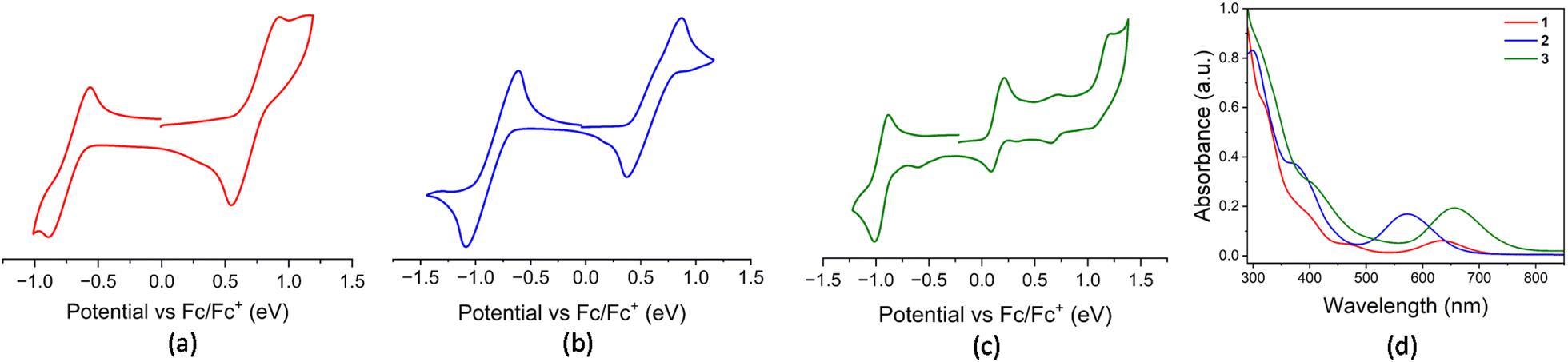 | ||
| Fig. 3 Cyclic voltammograms of 1 (a), 2 (b) and 3 (c) collected at 100 mV s−1; (d) combined UV-vis spectra of 1, 2 and 3. | ||
With an objective to gain more insightful information about the electronic transitions and structural characteristics of these Os–N,Se chelated complexes, UV-vis absorption spectroscopy was performed on complexes 1, 2, 3 and 4 in CH2Cl2. All these complexes exhibited intense absorption in the UV region and a notably distinct absorption pattern in the visible region. The strong absorption in the high-energy region appeared due to the π–π* transitions in ligand moieties. In addition, relatively weaker bands observed in the 300–400 nm range for both Os(II) and Os(III) species were ascribed to ligand-to-metal charge transfer (LMCT) transitions (Fig. 3(d) and S15). Notably, the paramagnetic Os(III)–N,Se chelated complexes exhibited broad absorptions extending into the lower-energy visible region, which can be assigned to the d(OsIII) → π*(ligand) metal-to-ligand charge transfer (MLCT). The introduction of an additional κ2-N,Se-L ligand in complex 2 caused a hypsochromic shift of this peak compared to complex 1. Furthermore, a bathochromic shift in the lower-energy absorption band was observed when going from complex 2 to 3, attributed to the presence of a different monodentate anionic ligand. Time-dependent DFT (TD-DFT) analysis shows that the transitions calculated at longer wavelengths for these complexes correspond to specific electronic excitations (Table S2). In complex 3, the transition at longer wavelength mainly arises from the excitations from β-HOMO−1 to β-LUMO, while in complex 2, analogous transitions from β-HOMO−2 and β-HOMO−3 to β-LUMO are observed in a relatively higher energy region.
It is worth noting that 2 can be regarded as electronically analogous to the [(L−)OsIII] tautomer of VIOs (vide supra, Chart 1)17c due to the absence of a non-innocent ligand. As a result, the osmium centre in 2 exhibits enhanced hardness associated with its higher oxidation state, while the sulfur atom in the osma-heterocycle of VIOs is replaced by a relatively softer selenium atom in 2. To examine the hemilability of the OsNCSe heterocycle in 2, its reactivity with free borane was investigated under various conditions. Thermolysis of 2 with four equivalents of [BH3·SMe2] at 60 °C changed its colour from deep green to orange and yielded 5 and 6 along with some air and moisture sensitive products (Scheme 2). The 31P{1H} NMR spectrum of 5 displayed a singlet at δ = 27.9 ppm which suggested the retention of the PPh3 unit. The appearance of four chemical shifts at δ = 59.9, 14.2, 4.6 and −15.2 ppm in the 11B{1H} NMR spectrum was attributed to the presence of four different boron environments. In addition to the aromatic region peaks, the room temperature 1H NMR spectrum of 5 unveiled three types of broad signals at δ = −11.66, −11.61 and −7.65 ppm in the upfield region. Additionally, mass spectrometry revealed isotopic distribution patterns at m/z 818.0868, consistent with the molecular formula [C28H31PN2Se2B4Os]. However, a comprehensive understanding could not be achieved until single-crystal X-ray diffraction analysis of complex 5 was performed.
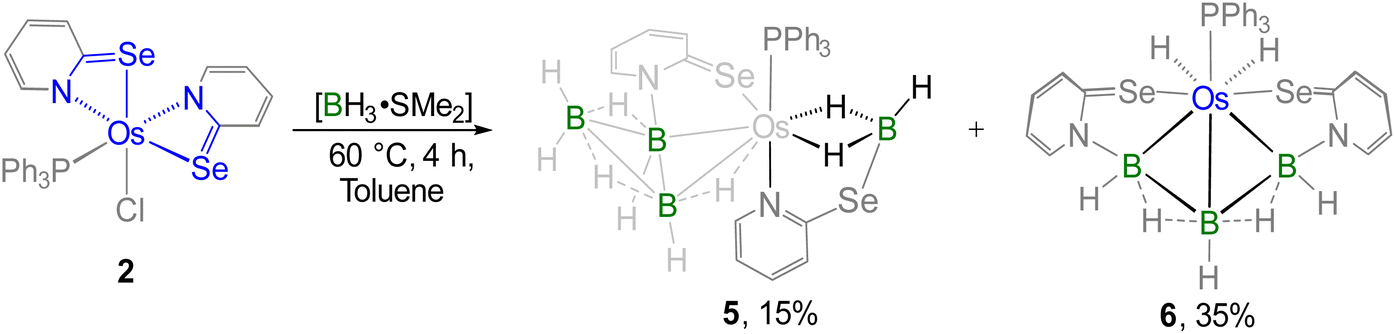 | ||
| Scheme 2 Cooperative borane activation utilizing Os(III)–N,Se-chelated complex 2. | ||
The X-ray diffraction study of 5 was carried out on an orange crystal obtained from the slow evaporation of hexane layered CH2Cl2 solution. As illustrated in Fig. 4, the osmium centre is coordinated to a substituted octatrihydridoborate unit, B3H7L, where its interaction with the metal fragment appears to differ from that observed in conventional TM–octatrihydridoborate complexes.23,24 Interestingly, the B2 atom is coordinated to osmium through an Os–H–B interaction, while a selenopyridinyl group bridges between Os1 and B1. The Os1–B1 bond length is shorter than that of Os1–B2, likely due to a secondary interaction mediated by the selenopyridinyl unit. In this context, the broad shielded 1H chemical shifts at δ = −7.65 ppm can be attributed to the presence of the Os–H–B unit. However, a definitive upfield signal characteristic of a B–H–B unit was not observed in the room-temperature 1H NMR spectrum. To probe this further, variable-temperature (VT) 1H NMR studies were performed. Upon lowering the temperature, two broad upfield resonances at δ = −0.39 and −2.64 ppm emerged, which can be attributed to two distinct B–H–B units. These findings indicate rapid exchange between the B–H–B and terminal B–H protons at room temperature. Note that a similar dynamic behaviour has also been reported for rhodatetraborane dihydride, [Rh(η2-B3H8)(H)2(PPh3)2].24a
 | ||
| Fig. 4 Molecular structure and labelling diagram of 5. Selected bond lengths (Å) of 5: Os1–B1 2.223(8), Os1–B2 2.306(8), Os1–B4 2.056(8), Os1–N2 2.132(5); B1–B2 1.758(12), B2–B3 1.771(13), B3–B1 1.862(12), and Se2–B4 2.001(8) (aromatic hydrogens are not shown for better clarity). | ||
On the other hand, the remaining coordination sites in complex 5 were occupied by one PPh3 ligand and a chelating ligand, [H3BL]− in a κ3-H,H,N bonding mode, leading to the formation of an Os–dihydridoborate moiety. The formation of this Os–dihydridoborate entity is proposed to result from the insertion of BH3 into the Os–Se bond of the κ2-N,Se-chelated ring in complex 5. The Os1–B4 separation aligns well with that observed in the Os–dihydridoborate complex, [Os(PPh3)(κ2-N,S-C5H4NS){κ3-H,H,S-H3BO(C5H4NS)}],17c but it is significantly shorter than the bond lengths typically found in TM–(σ-borane/borate) species.25,26 The bond separations of B4–Se2 and C12–Se2 are consistent with the typical ranges for B–Se and C–Se single bonds, respectively.27 Thus, complex 5 can be designated as an Os(dihydridoborate)(octatrihydridoborate) species, with the formulation [Os(PPh3)(κ3-H,H,N-BH3L){κ2-H,Se-B3H7L)}]. The formation of 5 likely proceeds via two concurrent steps: (i) the insertion of a preformed triborane fragment into the Os–N bond, resulting in the formation of the Os–octahydridotriborate unit, and (ii) the cooperative activation of two B–H bonds from BH3 through the cleavage of the Os–Se bond in another hemilabile OsNCSe metallacycle, which eventually furnishes the Os(dihydridoborate) unit. It is worth noting that this process constitutes a rare example of cooperative activation of borane in which both types of M–E bonds (E = N, Se) in hemilabile metallaheterocycle undergo cleavage.
Furthermore, complex 6 was obtained as a yellow crystalline solid after chromatographic separation. Notably, 6 could also be isolated in improved yield when [BH3·SMe2] was treated with the non-innocent ligand based Os–N,Se-chelated complex, 3 (Scheme 3). The 11B{1H} NMR spectrum of 6 displayed two distinct resonance signals at δ = 17.0 and −39.0 ppm, indicating the existence of two different boron environments. Furthermore, single phosphorus and selenium environments were identified by the singlet resonance at δ = 16.1 ppm in the 31P{1H} NMR spectrum and at δ = 234.8 ppm in the 77Se NMR spectrum, respectively. In the upfield region of the 1H NMR spectrum of 6, two non-equivalent hydride signals were detected at δ = −13.40 and −1.41 ppm, which presumably arise due to the presence of M–H and B–H–B protons, respectively. The ESI-MS data of 6 exhibited an isotopic pattern at m/z 805.0319 that aligns with the molecular formula [C28H15PN2Se2B3Os]. Although spectroscopic data in combination with mass spectrometry indicated the formation of an osmium borallyl species, structural confirmation was obtained by X-ray diffraction analysis of 6. The molecular structure of 6 reveals an Os–borallyl complex characterized by a primary η3-borallyl coordination of the [B3H5L2]2− fragment, along with secondary interactions involving the selenidopyridinyl selenium atom (Fig. S1). The spectroscopic and structural characteristics of 6 are consistent with those of a recently reported Os–borallyl complex, [(PPh3)(H)2Os(η5-B3H5(C5H4NS)2)].28 Similar to the analogous complex, the presence of two hydride ligands in 6 was confirmed by the sharp upfield resonance signal at δ = −13.40 ppm in the 1H NMR spectrum. The formation of 6 can be attributed to the insertion of the triborane unit facilitated by the ring opening of two osma-heterocycles.
 | ||
| Scheme 3 Cooperative activation of free and substituted boranes utilizing paramagnetic Os(III)–N,Se-chelated species, 2–3. | ||
As part of our ongoing research, we have studied the unconventional dual-site activation of both free and bulky boranes by the redox-active species, VIRu. (vide supra, Chart 1).17b Strikingly, the activation of free boranes led to the formation of [Ru(PPh3){κ3-H,S,S′-(NH2BSBH2N)(S2C7H4)2}], while a unique Ru-bis(dihydridoborate) complex was obtained from the cooperative activation of BH2Mes (Mes = (CH3)3C6H3).17b To investigate the influence of steric and electronic characteristics of substituted aryl groups of boranes on the cooperative activation process, we carried out reactions of complexes 2 and 3 with [BH2R·SMe2] and [BHR2·SMe2] (R = (CF3)2C6H3) (Scheme 3). Thermolysis of 2 with two equivalents of [BH2R·SMe2] generated 7 in 12% yield along with a few air-sensitive products, whereas complex 3 underwent decomposition under the same reaction conditions. The 31P{1H} NMR spectrum of 7 revealed a sharp singlet at δ = 22.3 ppm which suggested the retention of the PPh3 group. Similar to the 31P{1H} NMR spectrum, a single resonance was observed at δ = 27.8 ppm in the 11B{1H} NMR spectrum and at δ = −62.2 ppm in the 19F{1H} NMR spectrum. This suggested the presence of a highly symmetric geometry with chemically equivalent phosphorus, boron, and fluorine environments. Furthermore, the 1H NMR spectrum of 7 showed a broad upfield resonance at δ = −9.72 ppm, likely arising from the presence of an Os–H–B unit. Notably, no characteristic broad resonance corresponding to a terminal B–H proton was observed in the 1H NMR spectrum. However, the precise structure of 7 remained unresolved until solid-state X-ray diffraction analysis was performed on orange, needle-like crystals grown from a CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane mixture at −5 °C.
:hexane mixture at −5 °C.
As depicted in Fig. 5, complex 7 adopts a symmetrical core geometry featuring a mirror plane passing through the Se3–Os1–P1 fragment, which allows us to classify the molecule under Cs point group. The molecular structure of 7 consists of two five-membered {OsBNCSe} osmacycles fused via a single {OsB2Se} butterfly unit, where osmium and selenium were positioned at the hinge tips and boron atoms were located at the wing tips. Although we were unable to find any proper mechanistic evidence, we believe that the Se atom in the butterfly core is presumably generated from the C–Se bond cleavage of the selenopyridinyl moiety. Interestingly, complex 7 can be viewed as a tetraborane derivative, exhibiting structural and electronic resemblance with the Os–borallyl species, 6 and [(PPh3)(H)2Os(η5-B3H5(C5H4NS)2)].28 The average Os–B distance (2.263 Å) is elongated in comparison to the average Ru–B bond length of the analogous species, [(PPh3)Ru{κ3-H,S,S′-(NH2BSBH2N)(S2C7H4)2}],17b yet it aligns well with the typical distances observed in osmium-based σ-borane/borate species.29 Moreover, the average B–Se distance of 2.103 Å is slightly higher than that found in typical borane–selenate species.27 The formation of 7 can be described by the insertion of two BHR units via ring opening of both the Os–N bonds and subsequent formation of B–Se bonds.
 | ||
| Fig. 5 Molecular structure and labelling diagram of 7. Selected bond lengths (Å) and bond angles (°) of 7: Os1–Se1 2.4771(7), Os1–Se2 2.4705(7), Os1–Se3 2.5612(7), Os1–B1 2.289(7), Os1–B2 2.237(7), B1–Se3 2.113(7); B2–Se3 2.093(7) (aromatic hydrogens are not shown for better clarity). | ||
TM–borallyl complexes and their analogues are often unreactive due to the strong bonding between the borallyl entity and the metal centre. Additionally, these complexes typically feature strongly π-acidic or π-basic ligands, which further restrict their reactivity.28,30 However, in the case of complex 6, the osmium centre is coordinated to two hydride ligands and one PPh3 ligand which are relatively labile. To examine their lability, we performed prolonged thermolysis of 6 with an excess of [BH3·SMe2] that yielded complex 8 in moderate yield (Scheme 4). The 31P {1H} NMR analysis of 8 revealed no resonance peaks, indicating the loss of the PPh3 unit from 6. The 1H NMR spectrum of 8 at room temperature displayed broad resonance signals in the upfield region at δ = −9.83, −4.19, −2.79, −1.44, −1.29 and −0.54 ppm. These broad signals are due to the presence of either M–H–B or B–H–B protons. Additionally, in the 1H NMR spectrum, multiple sets of broad signals were observed in the range of δ = 3.74–2.66 ppm that correspond to terminal B–H protons while the 11B{1H} NMR spectrum shows peaks at δ = 12.2, 10.4, 8.9, −2.6, −7.2, −10.3, −11.5, −21.3, −22.4, and −28.1 ppm. The mass spectrometric data revealed an isotopic distribution pattern at m/z 593.0291. The comprehensive spectroscopic analysis suggests a highly asymmetrical nature of the complex. However, a definitive structure could not be established until X-ray diffraction analysis was conducted.
 | ||
| Scheme 4 Pyrolysis of Os–borallyl species [(PPh3)(H)2Os(η5-B3H5(C5H4NSe)2)], 6 with excess borane. | ||
The X-ray diffraction study was carried out on a suitable yellow crystal obtained from the slow evaporation of a 9/1 DCM/hexane solution of 8 at −4 °C. The solid-state X-ray diffraction study of 8 revealed an unusual geometry surrounding the osmium centre (Fig. 6). One part of the structure can be described as an Os–borallyl entity, wherein the triborane ligand [B3H5(C5H4NSe)2]2− coordinates to the osmium centre in an η3-fashion. The other part of the molecule resembles a pentaborane derivative, where the osmium atom occupies one of the vertices in the basal plane of a square pyramidal geometry. The core geometry of 8 can be best described as a fusion of a butterfly {OsB3} unit with a square pyramidal {OsB4} unit, with the osmium centre serving as the common vertex. The B–B and Os–B bond lengths in 8 fall within the range reported for metallaborane clusters.31,32 Notably, the average B–B bond length in the tetraborane analogue(1.83 Å) is slightly longer as compared to that of the pentaborane analogue (1.73 Å). However, the Os–B bond lengths in the osmatetraborane unit are comparatively longer than those in the osmatriborane unit. The formation of 8 probably proceeds via a two-step pathway: initial dissociation of PPh3 from complex 6, followed by the formation of {OsB4H8} via monoborane pyrolysis.32,33 Notably, a careful assessment of the crystallographic data revealed the presence of two different isomeric forms of 8 (Fig. S2 and S3). The primary distinction between these isomers lies in the relative orientation of the square-pyramidal {OsB4} unit with respect to the butterfly {OsB3} one. The co-existence of these isomers in solution accounts for the complexity observed in the 1H and 11B NMR spectra (Fig. S36–S40). Similar instances of isomeric co-existence have also been well observed in TM-complexes and metallaborane clusters.34
 | ||
| Fig. 6 Molecular structure and labelling diagram of 8. Selected bond lengths (Å) of 8: Os1–B1 2.416(3), Os1–B2 2.210(3), Os1–B3 2.268(3), B1–B2 1.713(4), and B1–B4 1.788(4) (aromatic hydrogens are not shown for better clarity). | ||
Notably, both complexes 6 and 8 feature a borallyl entity stabilized by heterocyclic ligand assisted secondary interactions involving the selenopyridinyl selenium atom. However, the bonding characterstics of other counterparts differ significantly from each other. To understand how this variation influences the bonding mode of the borallyl entity, we conducted DFT calculations. Molecular orbital (MO) analysis revealed that HOMO−37 of 6 exhibits extended bonding interaction across the triborane fragment (Fig. 7(a)), similar to the related complex [(PPh3)(H)2Os(η5-B3H5(C5H4NS)2)].28 Similarly, HOMO−26 of 8 displays a comparable interaction but with enhanced energy stabilization (Fig. 7(b)). The bonding interaction between osmium and the triborane unit is particularly pronounced in HOMO−27 of 6 (Fig. 7(c)) and HOMO−14 of 8 (Fig. 7(d)), with greater stabilization observed in the case of 6. This highlights the increased stability of the osmium borallyl framework in the presence of the PPh3 counterpart. This observation is also consistent with the lower charge accumulation on the wing-tip boron atoms of the borallyl unit in species 6. Moreover, HOMO−21 and HOMO−25 of 8 indicate the presence of substantial multi-centred bonding interactions within the {OsB4} square pyramidal core.
 | ||
| Fig. 7 (a) HOMO−37 of 6 (b) HOMO−26 of 8; (c) HOMO−27 of 6; (d) HOMO−14 of 8 (isovalue 0.004 [e per bohr3]1/2). | ||
Furthermore, we shifted our attention to the diamagnetic bis-κ2-N,Se-chelated species 4, which is analogous to recently reported [Os(PPh3)2(κ2-N,S-C5H4NS)2], 4S.17c Notably, complex 4S undergoes cooperative borane activation, where two inequivalent B–H activation events led to the formation of an Os(σ-borate)hydride species, 9 (Scheme 5).17c The primary difference between complexes 4 and 4S lies in the identity of their chalcogen atoms. Additionally, complex 4 is distinct from complexes 2 and 3 due to the different oxidation state of the osmium centre. To evaluate the impact of these electronic factors on metal–ligand cooperativity (MLC), complex 4 was subjected to the cooperative activation of both free and substituted borane. Treatment of 4 with one equivalent of [BH3·SMe2] led to the formation of complex 10 (Scheme 5). Interestingly, the 11B NMR spectrum of 10 displayed two signals at δ = 0.6 and −21.0 ppm, indicating the presence of two inequivalent boron environments. Likewise, the upfield region of the 1H NMR spectrum exhibited two broad signals at δ = −14.14 and −11.17 ppm, corresponding to two distinct Os–H–B units. Following the reaction, the appearance of two well-separated 31P{1H} NMR signals implied the emergence of an asymmetric phosphorus environment. Nonetheless, a detailed interpretation remained elusive until single-crystal X-ray diffraction analysis of complex 10 was conducted.
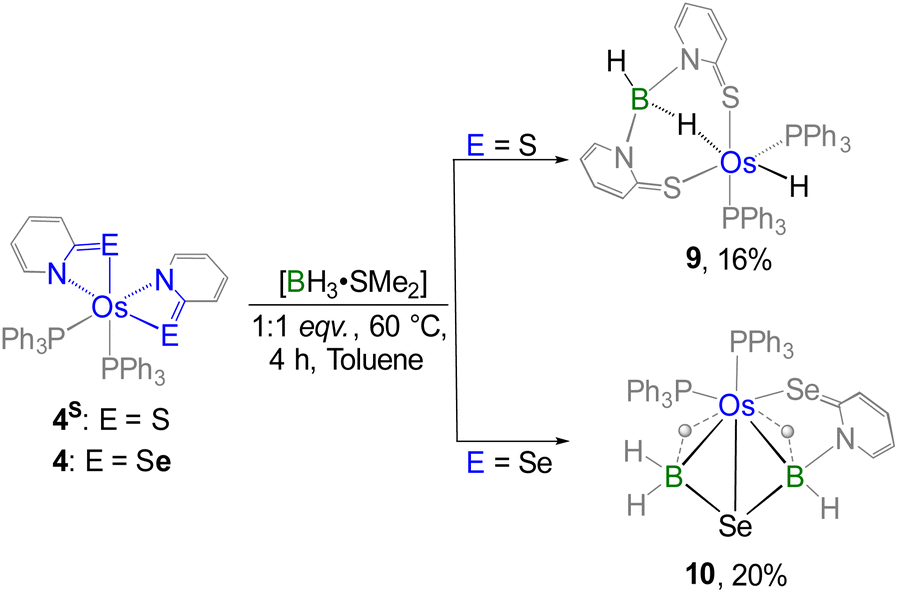 | ||
| Scheme 5 Cooperative activation of free borane utilizing diamagnetic Os(II)–N,E-chelated complexes (E = S, Se). | ||
As depicted in Fig. 8, the molecular structure of complex 10 reveals an octahedral geometry around the osmium centre, in which two PPh3 ligands, two bridging hydride ligands, and two selenium atoms are each positioned cis to one another. Similar to complex 7, complex 10 also contains a butterfly-shaped {OsB2Se} core and can thus be classified as a tetraborane derivative. The dihedral angle across the butterfly-shaped core in complex 10 (103.8°) is considerably smaller than that observed in the analogous complex 7 (112.2°) and the Os–borallyl species 6 (111.7°). This difference may be attributed to the presence of two inequivalent tetracoordinate borate moieties in complex 10, wherein one of the boron centres (B2) lacks coordination to a selenopyridinyl unit. Furthermore, the Os–Se and average B–Se bond distances are comparable in both complexes 7 and 10. Notably, the average Os–B bond length in complex 10 (2.376 Å) is significantly longer than that in complex 7 (2.263 Å), likely due to the presence of electron-withdrawing substituents in complex 7.
 | ||
| Fig. 8 Molecular structure and labelling diagram of 10. Selected bond lengths (Å) of 10: Os1–Se1 2.4950(5), Os1–Se2 2.529(2), Os1–B1 2.375(6), Os1–B2 2.380(5), B1–Se2 2.053(7); B2–Se2 2.078(6) (aromatic hydrogens and carbons are not shown for better clarity). | ||
The presence of the {OsB2Se} butterfly core with distinct ligand environments in both 7 and 10 prompted us to explore their bonding scenario. Molecular orbital (MO) analysis demonstrates that the HOMOs in both complexes are predominantly localized on the osmium centre and the selenium atom within the selenopyridinyl ring, whereas the LUMOs exhibit π-orbital delocalization across the pyridinyl ring. Importantly, the HOMO–LUMO energy gaps (ΔEHOMO–LUMO) are nearly identical for both complexes. Similarly, variations in ligand environments do not significantly influence the interaction between the osmium atom and the free selenium atom, as reflected by their similar bond distances and Wiberg bond index (WBI) values (Table S2). Natural bonding orbital (NBO) analysis further revealed the presence of three-centre two-electron (3c–2e) bonding interactions along the Os–H–B moieties (Fig. 9). In complex 7, the two boron atoms exhibit comparable natural charges, whereas in complex 10, the B1 atom shows higher charge accumulation than B2. This suggests that B1 possesses a more pronounced borate character, likely due to differences in its electronic environment.
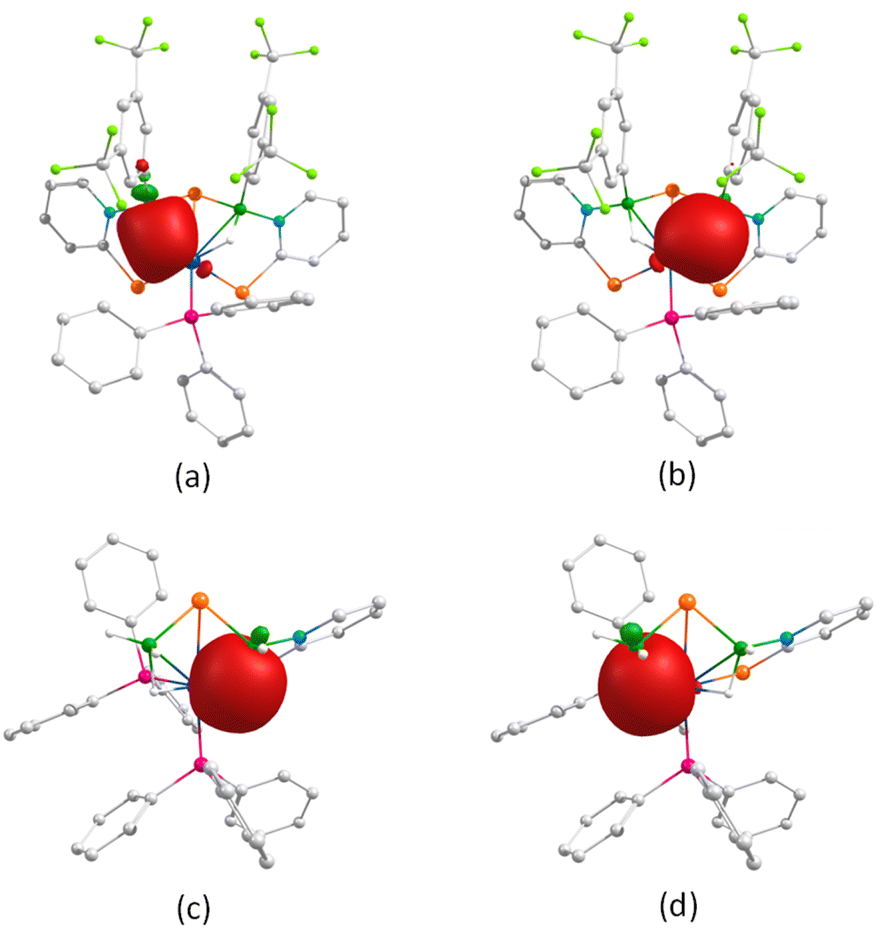 | ||
| Fig. 9 3c–2e bonding interaction along the Os–H–B unit of 7 (a and b) and 10 (c and d) (isovalue 0.004 [e per bohr3]1/2). | ||
On the other hand, complex 4 underwent decomposition when reacted with substituted boranes, [BH2R·SMe2] and [BHR2·SMe2]. To facilitate the cooperative activation of bulkier aryl-substituted boranes, we employed an alternative diamagnetic bis-κ2-N,S-chelated species, [Os(PPh3)2(κ2-N,S-C7H4NS2)2] (4mbz) in which the heterocyclic ligands impart a distinct electronic environment compared to its analogous species. Similar to the case of 4S, the cooperative B–H activation of free borane utilizing 4mbz resulted in the formation of the Os(σ-borate)hydride species, 9mbz.17c Building on this finding, the reaction of 4mbz with one equivalent of [BHR2·SMe2] at room temperature afforded complex 11 (Scheme 6). In contrast, reaction with [BH2R·SMe2] generated air and moisture-sensitive products in low yield. Complex 11 was isolated in 15% yield after chromatographic separation and further characterized by multinuclear spectroscopy, mass spectrometry and solid-state X-ray diffraction. The 11B{1H} NMR spectrum revealed a resonance peak at δ = 2.2 ppm that suggested the presence of a tetra-coordinated boron atom in a single environment. Furthermore, the 1H NMR spectrum of 11 exhibited a broad signal in the shielded region at δ = −5.51 ppm, which appeared relatively downfield compared to that in 10. The observed chemical shift is presumably due to the electron-withdrawing effect of the aryl substituents attached to boron. Notably, the appearance of two separate sharp signals in the 31P{1H} NMR spectrum implies that 4mbz experiences a reduction in symmetry following the reaction. Although the spectroscopic data suggested a probable activation of the B–H bond of BHR2, a detailed understanding of the structural characteristics of 11 could only be obtained through solid-state X-ray diffraction analysis.
 | ||
| Scheme 6 Cooperative activation of free and substituted boranes utilizing diamagnetic Os(II)–N,S-chelated complex 4mbz. | ||
As shown in Fig. 10, complex 11 can be identified as a κ2-N,S-chelated Os(σ-borate) complex which can be formulated as [Os(PPh3)2(κ2-N,S-C7H4NS2)(κ3-H,S,S′-HBR2(C7H4NS2))]. The molecular structure of 11 exhibited a distorted octahedral geometry around the osmium centre with the retention of two PPh3 units and one OsNCS metallacycle from 4mbz. Additionally, the formation of a nonplanar six-membered osma-heterocycle (OsHBNCS) can be attributed to the hemilabile ring opening of an Os–N bond, followed by the capture of a B–H bond of BHR2. The Os–B separation in complex 11 is considerably larger than that found in reported Os(σ-borane) complexes29 as well as other σ-borate complexes of group-8 TM.17,25a It is noteworthy that one of the osma-heterocycles (OsNCS) remained intact after the reaction, and its bite angle is comparable to that of other OsNCS metallacycles.17c However, we believe that the steric hindrance caused by the two aryl groups on the boron atom prevented the B–H activation of the second BHR2 unit. Indeed, this can be considered as one of the rarest examples of cooperative activation involving a sterically demanding di-substituted borane.
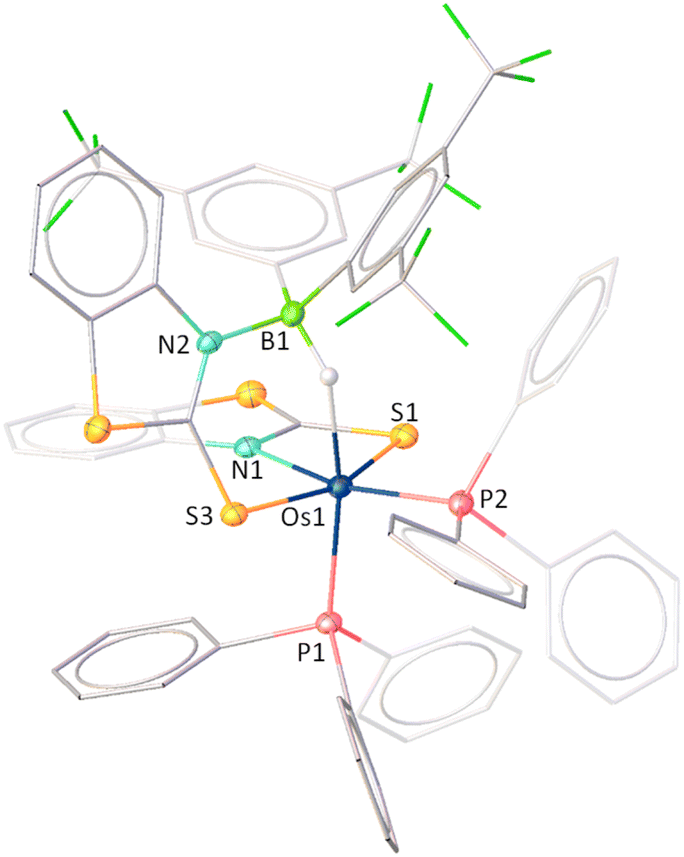 | ||
| Fig. 10 Molecular structure and labelling diagram of 11. Selected bond lengths (Å) and bond angles (°) of 11: Os1–S1 2.441(3), Os1–S3 2.339(3), Os1–P1 2.309(3), Os1–P2 2.314(3), Os1–B1 3.001, and N1–Os1–S1 67.3(2) (aromatic hydrogens are not shown for better clarity). | ||
DFT calculations offered valuable insights into the energetics of the cooperative B–H activation of BR2H as well as the bonding nature in complex 11. The observed negative Gibbs free energy change (ΔG = −12.6 kcal mol−1) for this reaction indicates that the sterically hindered borane activation by 4mbz is thermodynamically favourable. Molecular orbital (MO) analysis revealed that HOMO is primarily localized on the metal centre and the mercaptobenzothiazolyl sulphur atoms, while the LUMO is mainly delocalized over the aryl substituents of boron with only marginal involvement of the metal centre. A notable HOMO–LUMO energy gap of 3.379 eV was identified for 11. Natural bonding orbital (NBO) analysis further confirmed a strong donor–acceptor interaction between the σ(B–H) orbital and the Os centre in 11 (Fig. 11, left). Second-order perturbation theory estimated a substantial stabilization energy of 43.9 kcal mol−1, emphasizing the strength of this interaction. Additionally, the σ-borate unit in 11 was found to exhibit bond critical points (BCPs) along the Os–H and B–H bonds (Fig. 11, right).
 | ||
| Fig. 11 Donor–acceptor interaction among the σ(B–H) orbital and Os centre in 11 (left); contour-line diagram of the Laplacian of the electron density along Os–H–B planes in 11 (right). | ||
Conclusions
In this study, we have developed various Os(III)-based 1,3-N,Se-chelated complexes as model systems for investigating metal–ligand cooperativity. By systematically tuning the electronic characteristics of both the metal and the ligand frameworks, we modulated the hemilability of osma-heterocycle motifs. The modes of electron localization within these paramagnetic species were thoroughly characterized using spectroscopic, electrochemical, and photophysical techniques. To evaluate the impact of these tailored electronic characteristics, we investigated the cooperative activation of free borane. The cooperative activation of free borane employing the bis-1,3-N,Se-chelated Os(III) complex (2) resulted in the formation of a unique Os(dihydridoborate)(octatrihydridoborate) complex. This transformation proceeds via cleavage of both the Os–Se and Os–N bonds of two different metallacycles. In contrast, when employing complex 3, the activation was dominated by selective Os–N bond cleavage, highlighting the sensitivity of the cooperative activation pathway to the electronic environment of the complex. We further examined the distinct activation pattern of sterically hindered boranes using various paramagnetic and diamagnetic bis-1,3-N,E-chelated complexes (E = S, Se). The mono- and di-substituted hydroboranes led to the formation of Os(σ-borate) species through cooperative B–H bond activation. Interestingly, with a sterically demanding di-aryl substituted borane, only one metallacycle unit underwent activation, while the second osma-heterocycle remained intact, presumably due to steric hindrance of aryl substituents. This study provides valuable insights into metal–ligand cooperativity and opens the way for the development of unique transition metal–boron complexes with potential applications in catalysis.Author contributions
F. Assanar and S. Gayen have executed the experimental synthesis, characterization and analysis of the data. S. Gayen has performed the theoretical calculations. D. K. Patel has performed the X-ray diffraction analysis. All authors have contributed to the preparation of the manuscript. S. Ghosh and T. Pradeep have supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc06346a.CCDC 2481059 (1), 2481057 (2), 2466501 (4), 2481058 (5), 2466499 (6) 2466503 (7), 2466500 (8), 2472949 (10) and 2472945 (11) contain the supplementary crystallographic data for this paper.35a–i
Acknowledgements
S. Ghosh acknowledges the support of the SERB, India, Grant No. CRG/2023/000189. T. Pradeep thanks SERB for funding through Grant No. SPR/2021/000439 and the JC Bose Fellowship. We thankfully acknowledge the support from the CoE on Molecular Materials and Functions under the IoE scheme, IIT Madras for the single crystal XRD facility. F. A. thanks UGC, S. G. thanks CSIR and D. K. P. thanks PMRF (Grant No. SB22230356CYPMRF008224) for research fellowships. The computational facility of IIT Madras is gratefully acknowledged.Notes and references
- (a) R. Peters, Cooperative Catalysis, Wiley-VCH, Weinheim, 2015 CrossRef; (b) J. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed; (c) L. Buchwald and D. Milstein, Ligand Design in Metal Chemistry: Reactivity and Catalysis, John Wiley & Sons, 2016 Search PubMed.
- (a) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef CAS; (b) B. Chatterjee, W. C. Chang, S. Jena and C. Werle, ACS Catal., 2020, 10(23), 14024–14055 CrossRef CAS; (c) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed.
- (a) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; (b) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed; (c) M. Stradiotto and R. J. Lundgren, Ligand Design in Metal Chemistry, Wiley Online Library, 2016 CrossRef.
- (a) M. Bassetti, Eur. J. Inorg. Chem., 2006, 4473–4482 CrossRef CAS; (b) P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136(4), 1367–1380 CrossRef CAS PubMed; (c) F. Forster and M. Oestreich, Metal–Ligand Cooperative Si–H Bond Activation in Organosilicon Chemistry–Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 115–130 Search PubMed; (d) L. Omann, C. D. F. Konigs, H. F. T. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50(5), 1258–1269 CrossRef CAS PubMed; (e) S. P. Cronin, J. M. Strain, M. S. Mashuta, J. M. Spurgeon, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2020, 59(7), 4835–4841 CrossRef CAS PubMed.
- (a) K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC; (b) C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC; (c) Q. Liang, H. A. G. Mayerstein and D. Song, Organometallics, 2023, 42(9), 816–824 CrossRef CAS; (d) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119(8), 10393–10402 CrossRef CAS PubMed; (e) S. Zhang, X. Zhai, Y. Song, L. Feng, C. H. Tung and W. Wang, Organometallics, 2021, 40(11), 1692–1698 CrossRef CAS; (f) M. G. Cameo, A. Urriolabeitia, E. Barrenas, V. Passarelli, J. J. Pérez-Torrente, A. D. Giuseppe, V. Polo and R. Castarlenas, ACS Catal., 2021, 11(12), 7553–7567 CrossRef; (g) B. Español-Sánchez, J. Moradell, M. Galiana-Cameo, E. Barrenas, J. J. Pérez-Torrente, V. Passarelli and R. Castarlenas, Angew. Chem., Int. Ed., 2025, 64, e202507424 CrossRef PubMed.
- (a) G. R. Owen, Chem. Soc. Rev., 2012, 41, 3535–3546 RSC; (b) G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2016, 45, 1065–1079 RSC; (c) J. A. Zurakowski, B. Stadler, M. W. Drover and M. R. Crimmin, Angew. Chem., Int. Ed., 2025, 64, e202512684 CrossRef CAS PubMed.
- (a) W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134(11), 5080–5082 CrossRef CAS PubMed; (b) A. M. Poitras, S. E. Knight, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Angew. Chem., Int. Ed., 2018, 57(6), 1497–1500 CrossRef CAS PubMed; (c) W. H. Harman, T. P. Lin and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53(4), 1081–1086 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44(8), 588–602 CrossRef CAS PubMed; (b) R. H. Morris, Acc. Chem. Res., 2015, 48(5), 1494–1502 CrossRef CAS PubMed; (c) M. Rauch, S. Kar, A. Kumar, L. Avram, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2020, 142(34), 14513–14521 CrossRef CAS PubMed; (d) Y. Liang, J. Luo, Y. D. Posner and D. Milstein, J. Am. Chem. Soc., 2023, 145(16), 9164–9175 CrossRef CAS PubMed.
- (a) H. Li, M. Fan and Q. Liu, J. Am. Chem. Soc., 2024, 146(39), 26649–26656 CrossRef CAS PubMed; (b) J. J. Gair, Y. Qiu, R. L. Khade, N. H. Chan, A. S. Filatov, Y. Zhang and J. C. Lewis, Organometallics, 2019, 38(7), 1407–1412 CrossRef CAS; (c) H. Li, T. P. Goncalves, D. Lupp and K. W. Huang, ACS Catal., 2019, 9(3), 1619–1629 CrossRef CAS.
- (a) V. Lyaskovskyy and B. d. Bruin, ACS Catal., 2012, 2(2), 270–279 CrossRef CAS; (b) D. L. Broere, R. Plessius and J. I. V. Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC; (c) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- (a) C. Erken, A. Kaithal, S. Sen, T. Weyhermuller, M. Holscher, C. Werle and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed; (b) B. Wang, C. Rong, M. Lei, S. Liu and F. D. Proft, Inorg. Chem., 2023, 62(19), 7366–7375 CrossRef CAS PubMed; (c) H. Song, K. Ye, P. Geng, X. Han, R. Liao, C.-H. Tung and W. Wang, ACS Catal., 2017, 7(11), 7709–7717 CrossRef CAS; (d) M. Pang, C. Wu, X. Zhuang, F. Zhang, M. Su, Q. Tong, C.-H. Tung and W. Wang, Organometallics, 2018, 37(9), 1462–1467 CrossRef CAS; (e) X. Zhai, M. Pang, L. Feng, J. Jia, C. H. Tung and W. Wang, Chem. Sci., 2021, 12, 2885–2889 RSC.
- (a) M. R. Elsby and R. T. Baker, Chem. Commun., 2019, 55, 13574–13577 RSC; (b) M. A. Nesbit, D. L. Suess and J. C. Peters, Organometallics, 2015, 34(19), 4741–4752 CrossRef CAS; (c) S. N. MacMillan, W. H. Harman and J. C. Peters, Chem. Sci., 2014, 5, 590–597 RSC; (d) S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed., 2021, 60, 14272–14294 CrossRef CAS PubMed; (e) L. A. Grose and D. Willcox, Chem. Commun., 2023, 59, 7427–7430 RSC.
- (a) J. F. C. Perez, F. L. Kirlin, E. F. Reynolds, C. E. A. Jarczyk, B. T. Joseph, J. M. Keith and A. R. Chianese, ACS Catal., 2024, 14(21), 16497–16507 CrossRef CAS PubMed; (b) P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136(4), 1367–1380 CrossRef CAS PubMed; (c) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53(33), 8722–8726 CrossRef CAS PubMed; (d) B. L. Ramirez and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 5396–5407 CrossRef CAS PubMed; (e) M. Hasenbeck, T. Müller and U. Gellrich, Catal. Sci. Technol., 2019, 9, 2438–2444 RSC.
- (a) K. Fujita, N. Tanino and R. Yamaguchi, Org. Lett., 2007, 9(1), 109–111 CrossRef CAS PubMed; (b) A. M. Royer, T. B. Rauchfuss and D. L. Gray, Organometallics, 2010, 29, 6763–6768 CrossRef CAS.
- (a) M. W. Drover, L. L. Schafer and J. A. Love, Angew. Chem., Int. Ed., 2016, 55(9), 3181–3186 CrossRef CAS PubMed; (b) M. W. Drover, J. A. Love and L. L. Schafer, J. Am. Chem. Soc., 2016, 138(27), 8396–8399 CrossRef CAS PubMed; (c) N. M. Leeb, M. W. Drover, J. A. Love and L. L. Schafer, Organometallics, 2018, 37(24), 4630–4638 CrossRef CAS.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137(40), 12808–12814 CrossRef CAS PubMed.
- (a) Md. Zafar, R. Ramalakshmi, A. Ahmad, P. K. S. Antharjanam, S. Bontemps, S. S. Etienne and S. Ghosh, Inorg. Chem., 2021, 60(2), 1183–1194 CrossRef CAS PubMed; (b) M. Zafar, A. Ahmad, S. Saha, R. Rongala, T. Roisnel and S. Ghosh, Chem. Sci., 2022, 13, 8567–8575 RSC; (c) S. Gayen, F. Assanar, S. Shyamal, D. P. Dorairaj and S. Ghosh, Chem. Sci., 2024, 15, 15913–15924 RSC.
- (a) M. Zafar, R. Ramalakshmi, K. Pathak, A. Ahmad, T. Roisnel and S. Ghosh, Chem. Eur J., 2019, 25, 13537–13546 CrossRef CAS PubMed; (b) A. Ahmad, S. Saha, M. Zafar, T. Roisnel, P. Ghosh and S. Ghosh, Eur. J. Org Chem., 2023, 989, e202201283 CrossRef.
- (a) S. Saha, A. Haridas, F. Assanar, C. Bansal, P. K. S. Antharjanam and S. Ghosh, Dalton Trans., 2022, 51, 4806–4813 RSC; (b) S. Saha, F. Assanar, A. Ahmad, A. Bains, S. Nagendran and S. Ghosh, Organometallics, 2024, 43(7), 718–725 CrossRef CAS.
- (a) R. E. DeSimone, J. Am. Chem. Soc., 1973, 95(19), 6238–6244 CrossRef CAS; (b) P. Singh, B. Sarkar, M. Sieger, M. Niemeyer, J. Fiedler, S. Zalis and W. Kaim, Inorg. Chem., 2006, 45(12), 4602–4609 CrossRef CAS PubMed.
- (a) P.-S. Kuhn, G. E. Buchel, K. K. Jovanovic, L. Filipovic, S. Radulovic, P. Rapta and V. B. Arion, Inorg. Chem., 2014, 53, 11130–11139 CrossRef CAS PubMed; (b) F. Chen, G.-F. Wang, Y.-Z. Li, X.-T. Chen and Z.-L. Xue, Inorg. Chem. Commun., 2012, 21, 88–91 CrossRef CAS.
- K. W. Given, S. H. Wheeler, B. S. Jick, L. J. Maheu and L. H. Pignolet, Inorg. Chem., 1979, 18, 1261–1266 CrossRef CAS.
- (a) N. N. Greenwood, J. D. Kennedy, M. T. Pett and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1985, 2397–2406 RSC; (b) M. Bown, X. L. R. Fontaine, N. N. Greenwood, P. MacKinnon, J. D. Kennedy and M. T. Pett, J. Chem. Soc., Dalton Trans., 1987, 2781–2787 RSC.
- (a) F. Diaw-Ndiaye, P. J. S. Miguel, R. Rodríguez and R. Macías, Molecules, 2023, 28(18), 6462 CrossRef CAS PubMed; (b) A. Ahmad, S. Saha, M. Zafar, M. Cordier and S. Ghosh, Eur. J. Inorg. Chem., 2023, 26, e202300196 CrossRef CAS.
- (a) K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260–1273 CrossRef CAS PubMed; (b) K. Saha, R. Ramalakshmi, S. Gomosta, K. Pathak, V. Dorcet, T. Roisnel, J.-F. Halet and S. Ghosh, Chem. Eur J., 2017, 23, 9812–9820 CrossRef CAS PubMed; (c) S. Gayen, S. Shyamal, S. Mohapatra, P. K. S. Antharjanam and S. Ghosh, Chem. Eur J., 2024, 30, e202302362 CrossRef CAS PubMed; (d) M. R. St.-J. Foreman, A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 4446–4450 CrossRef CAS.
- (a) C. Lenczyk, D. K. Roy, J. Nitsch, K. Radacki, F. Rauch, R. D. Dewhurst, F. M. Bickelhaupt, T. B. Marder and H. Braunschweig, Chem. Eur J., 2019, 25, 13566–13571 CrossRef CAS PubMed; (b) R. N. Perutz, S. S. Etienne and A. S. Weller, Angew. Chem., Int. Ed., 2021, 60, 2–24 CrossRef; (c) C. N. Muhoro, X. He and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121(21), 5033–5046 CrossRef CAS.
- (a) D. Auerhammer, M. Arrowsmith, R. D. Dewhurst, T. Kupfer, J. Bohnke and H. Braunschweig, Chem. Sci., 2018, 9, 2252–2260 RSC; (b) A. Ahmad, S. Gayen, S. Mishra, Z. Afsan, S. Bontemps and S. Ghosh, Inorg. Chem., 2024, 63(7), 3376–3382 CrossRef CAS PubMed.
- S. Gayen, S. Shyamal, F. Assanar and S. Ghosh, Organometallics, 2024, 43(22), 2935–2943 CrossRef CAS.
- (a) M. L. Buil, J. J. F. Cardo, M. A. Esteruelas, I. Fernandez and E. Onate, Organometallics, 2015, 34(3), 547–550 CrossRef CAS; (b) J. C. Babon, M. A. Esteruelas, I. Fernandez, A. M. Lopez and E. Onate, Inorg. Chem., 2018, 57(8), 4482–4491 CrossRef CAS PubMed; (c) N. Arnold, S. Mozo, U. Paul, U. Radius and H. Braunschweig, Organometallics, 2015, 34(24), 5709–5715 CrossRef CAS.
- X. Lei, M. Shang and T. P. Fehlner, Organometallics, 1998, 17(8), 1558–1563 CrossRef CAS.
- (a) S. Shyamal, D. Chatterjee, K. Kar and S. Ghosh, Inorg. Chem., 2024, 63(46), 21838–21848 CrossRef CAS PubMed; (b) K. Kar, S. Kar and S. Ghosh, Chem. Sci., 2024, 15, 4179–4186 RSC.
- (a) S. K. Bose, K. Geetharani, B. Varghese and S. Ghosh, Inorg. Chem., 2010, 49(14), 6375–6377 CrossRef CAS PubMed; (b) K. Geetharani, B. S. Krishnamoorthy, S. Kahlal, S. M. Mobin, J.-F. Halet and S. Ghosh, Inorg. Chem., 2012, 51(19), 10176–10184 CrossRef CAS PubMed; (c) R. Borthakur, K. Saha, S. Kar and S. Ghosh, Coord. Chem. Rev., 2019, 399, 213021 CrossRef CAS; (d) S. Kar, A. N. Pradhan and S. Ghosh, Coord. Chem. Rev., 2021, 436, 213796 CrossRef CAS.
- (a) S. Ghosh, B. C. Noll and T. P. Fehlner, Dalton Trans., 2008, 371–378 RSC; (b) S. Sahoo, R. S. Dhayal, B. Varghese and S. Ghosh, Organometallics, 2009, 28(5), 1586–1589 CrossRef CAS; (c) S. Ghosh, B. C. Noll and T. P. Fehlner, Angew. Chem., Int. Ed., 2005, 44(19), 2916–2918 CrossRef CAS PubMed; (d) R. S. Anju, D. K. Roy, K. Geetharani, B. Mondal, B. Varghese and S. Ghosh, Dalton Trans., 2013, 42, 12828–12831 RSC.
- (a) J. R. Shapley, J. T. Park, M. R. Churchill, C. Bueno and H. J. Wasserman, J. Am. Chem. Soc., 1981, 103, 7385–7387 CrossRef CAS; (b) U. Kölle, J. Kossakowski, N. Klaff, L. Wesemann, U. Englert and G. E. Herberich, Angew. Chem., Int. Ed., 1991, 30, 690–691 CrossRef; (c) S. Ghosh, X. Lei, C. L. Cahill and T. P. Fehlner, Angew. Chem., Int. Ed., 2000, 39, 2900–2902 CrossRef CAS.
- (a) CCDC 2481059: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p8r5x.; (b) CCDC 2481057: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p8r3v; (c) CCDC 2466501: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nslkm; (d) CCDC 2481058: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p8r4w; (e) CCDC 2466499: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nslhk; (f) CCDC 2466503: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nslmp; (g) CCDC 2466500: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nsljl; (h) CCDC 2472949: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p09kl; (i) CCDC 2472945: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p09fg.
| This journal is © The Royal Society of Chemistry 2026 |