
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-surface synthesis of nitrogen-doped nanographenes assisted by self-assembly
Maria
Tenorio†
 a,
Cesar
Moreno
*b,
Jesús
Castro-Esteban‡
c,
Manuel
Vilas-Varela
c,
Diego
Peña
*cd and
Aitor
Mugarza
*ae
a,
Cesar
Moreno
*b,
Jesús
Castro-Esteban‡
c,
Manuel
Vilas-Varela
c,
Diego
Peña
*cd and
Aitor
Mugarza
*ae
aCatalan Institute of Nanoscience and Nanotechnology (ICN2) CSIC, The Barcelona Institute of Science and Technology, Campus UAB Bellaterra, Barcelona, 08193, Spain. E-mail: aitor.mugarza@icn2.cat
bDepartamento de Ciencias de la Tierra y Física de la Materia Condensada, Universidad de Cantabria, Santander, 39005, Spain. E-mail: cesar.moreno@unican.es
cCentro de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidad de Santiago de Compostela, Santiago de Compostela, 15702, Spain. E-mail: diego.pena@usc.es
dOportunius, Galician Innovation Agency (GAIN), Santiago de Compostela, 15702, Spain
eICREA Institució Catalana de Recerca i Estudis Avançats, Lluis Companys 23, Barcelona, 08010, Spain
First published on 11th December 2025
Abstract
Despite the vast literature on in-solution synthesized heterocycle-based nanographenes, examples of their on-surface synthesized counterparts remain remarkably scarce. A major limitation is related to the stability of heterogroups throughout the thermal reaction steps. In this study, we show how the supramolecular steric hindrance arising from densely packed, saturated monolayers of the molecular precursor, suppresses Ullmann polymerization and significantly reduces the temperature required for internal cyclodehydrogenation from 300 °C down to 140 °C. This alternative route leads to the formation of self-assembled islands of a prochiral chloro-substituted nitrogen-doped ovalene derivative, offering a promising strategy for the synthesis of doped nanographenes under mild thermal conditions. Such conditions are crucial for preserving sensitive functional groups or heteroatoms and enable the integration of these reactions into more complex synthetic sequences.
Introduction
Nanographenes, also known as graphene quantum dots or molecular graphenes, are laterally confined zero dimensional systems formed by sp2 hybridized carbons that have long been known to exhibit specific optical,1–4 electronic5 and optoelectronic6,7 properties depending on their size, edge topology and heteroatom content.6 Solution-based synthesis has allowed the execution of most of the aforementioned applications, comprising a vast literature in the field of synthesis of nanographenes species.8 Recently, surface-assisted methods have emerged as an interesting technique in order to synthesize insoluble and more reactive species. The synthesis under UHV conditions enables the local, atomic scale characterization of structural features such as chirality,9–11 or electronic and magnetic properties.12–17 However, the motivation to synthesize more and more complex species is limited by the thermal activation of reaction steps in on-surface synthesis (OSS), especially when heteroatoms play a role in the reaction route. The most established reaction to induce the formation of planar nanographenes is via cyclodehydrogenation (CDH) from non-planar precursors,18 a reaction that is triggered at high temperatures and consequently is often accompanied by uncontrolled side reactions.19 Likewise, this toll has an impact on the integrity of embedded heteroatoms for functionalization and doping purposes,20,21 as many are unstable, but able to withstand the high temperatures of thermally induced reactions.22 This is the reason why current literature shows examples limited to systems where the heteroatoms are at the core of the nanographene,23,24 with scarce reported cases with low yield of edge-doped nanographenes.25,26The set of controllable parameters typically explored in OSS to steer the formation of desired products27 are the choice of precursor and catalytic metal, and the type and temperature of reaction. Kinetic or steric factors such as the deposition and annealing rate, or the self-assembly of molecular precursors are seldom considered in the synthetic method as parameters to control and guide reaction routes. One of the most studied routes for the formation of graphene nanostructures considers 10,10′-dibromo-9,9′-bianthryl derivatives19,22,28–32 deposited on Au(111), and uses a two-step process based on the Ullmann polymerization and internal CDH reactions.
Here, we demonstrate a new reaction path for a 10,10′-dibromo-9,9′-bianthryl derivative where the Ullmann coupling has been effectively inhibited, promoting the formation of a prochiral, chloro-substituted nitrogen-doped ovalene derivative via the direct CDH of monomers. At this high coverage, the steric hindrance among monomers that are self-assembled in large islands and locally saturate surface terraces impedes the Ullmann cross-coupling and lowers the activation barrier for the internal CDH. Tracking the reaction path by combining Scanning Tunneling Microscopy (STM) with X-ray Photoelectron Spectroscopy (XPS), we reveal that the intrinsic chirality of the molecular precursors and their organization on the surface, triggered by the highly packed organization, also play a pivotal role in the yield towards the final product. We focus on the effect that chiral-dependent molecular interactions have in guiding the chemical route towards the anticipated intramolecular reactions. Once the nitrogen-doped nanographenes are formed, these are stabilized and self-assembled via N⋯H and Cl⋯H interactions. Our results provide insight to inspire new on-surface strategies towards the synthesis of more reactive species carrying unstable functional groups.
Experimental
The molecular building block under study is 10,10′-dibromo-2,2′-bis(2-chloropyrimidin-5-yl)-9,9′-bianthracene (M in Fig. 1), a pyrimidine-substituted bianthracene derivative that had been synthesized in solution following a procedure reported previously.33 Monomers were sublimated from a commercial Dodecon Knudsen type effusion cell on an atomically clean Au(111) surface at rates of ca 0.1 ML min−1. The Au(111) was previously cleaned by repeated cycles of sputtering (2 × 10−6 mbar of Ar+ at 1 KV) and annealing (470 °C) for 10 minutes each. The sample was prepared and post-annealed in a specific UHV preparation chamber (<1 × 10−9 mbar), and then transferred to the attached analysis UHV chamber, equipped with a Createc LT-STM (base pressure <1 × 10−10 mbar, and T = 4.5 K) for topography characterisation. During the annealing process, the temperature was directly monitored using a thermocouple spot-welded to the sample holder. Samples characterised with XPS were prepared and post-annealed in a UHV preparation chamber (5 × 10−9 mbar) attached to the XPS analysis chamber (1 × 10−9 mbar and RT). The temperature at each step of post-annealing was measured with an infrared pyrometer previously calibrated against a thermocouple fixed to the sample holder. XPS spectra were acquired with a SPECS Phoibos 150 hemispherical energy analyzer using a monochromatic Al Kα X-ray source at 1486.6 eV. XPS spectra were referenced to the Fermi level, and fitted with a Fermi–Dirac distribution at the sample temperature. The fitting results are shown in the SI.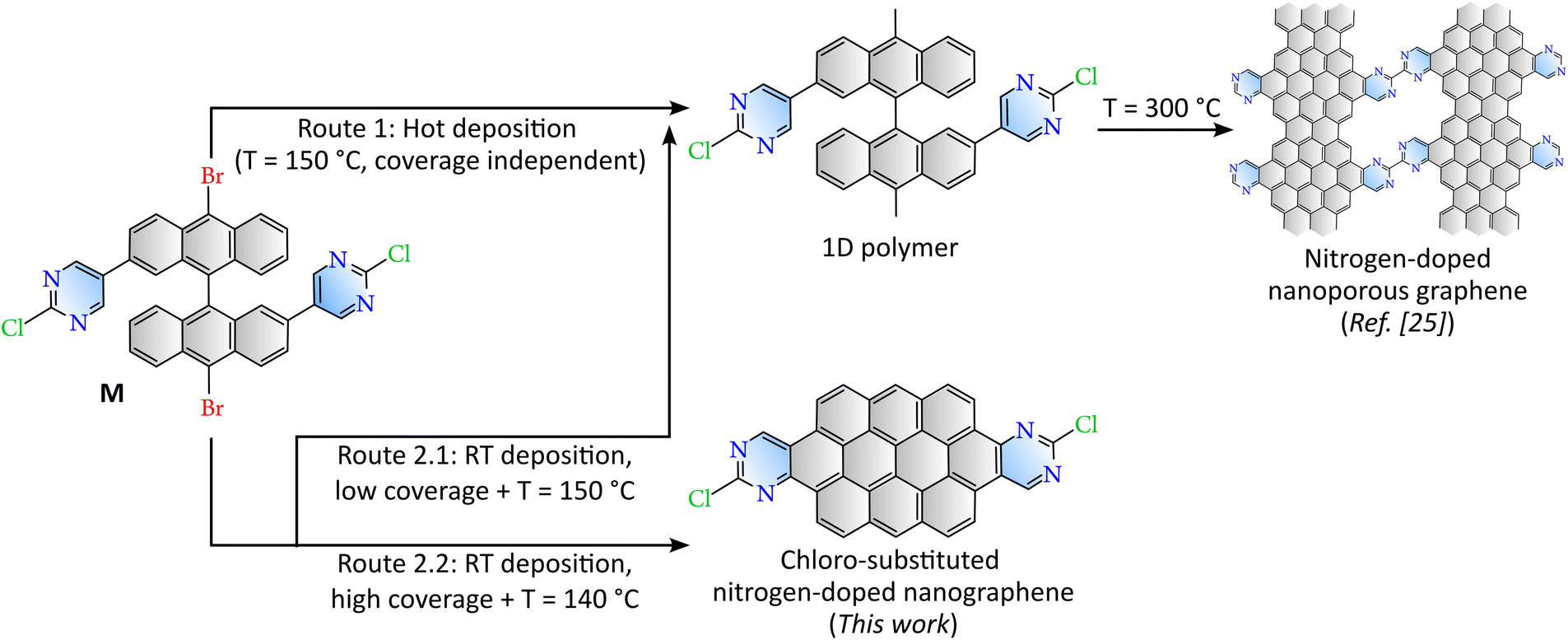 | ||
| Fig. 1 Schematic representation of the on-surface synthesis of different graphene-based nanoarchitectures depending on the deposition conditions from the molecular precursor M on Au(111). Pyrimidine functional substituents have been colored in blue, whereas the pure graphene-based molecule is grey. | ||
Results and discussion
The different graphene-based nanoarchitectures derived from the molecular precursor M under specific sublimation conditions are depicted in Fig. 1. Reaction route 1 (ref. 22) takes place when M is deposited on Au(111) held at 100–150 °C. This path is independent of coverage, since Ullmann polymerization is expected to occur as precursors land on the surface at the reaction temperature. When the precursor is deposited at room temperature (RT), the route (2.1 or 2.2) becomes coverage dependent. Well below 1 ML (route 2.1), post-annealing to 150 °C results on the same Ullmann polymerization as that found for the hot deposition in route 1, as described previously.22 On the other hand, at a coverage close to 1 ML, the reaction route 2.2 studied in this work takes place, giving rise to the growth of nitrogen-doped nanographenes at significantly reduced temperatures.Route 2.2 has been tracked by STM after sequential postannealing stages. Topographic images representative of the corresponding steps are displayed in Fig. 2. Molecular precursors, which are characterized by an intrinsic axial chirality, self-assemble at room temperature in homochiral domains, (phase A in Fig. 2a. The axial chirality is further described in Section 1 of the SI).22 The surface annealed at 100 °C still hosts well-ordered monomeric self-assembled islands, in stark contrast with the complete polymerization demonstrated recently by following paths 1 or 2.1.22 Furthermore, in the clean surface regions surrounding the large islands composed of close-packed molecules, randomly distributed monomers interact (see also Fig. S2 for comparison), revealing a completely different scenario to what is observed when polymers are formed. At this temperature stage, a new phase (phase B in Fig. 2b) coexists within the island with the as-deposited homochiral phase A. The molecular model that fits best to the topographic image is that of a racemic mixture of monomers (Fig. S3). We attribute this phase B to debrominated monomeric chains that are stabilized by coordination to native gold adatoms of the substrate.34,35
 | ||
| Fig. 2 STM topography characterisation of the on-surface formation of nitrogen-doped nanographenes on Au(111) (path 2.2 in Fig. 1) (a) top: overview of the as-deposited molecules. Bottom: close-up STM image of the A phase. IV conditions: (overview) 275 nm × 275 nm, 40 pA, 1.5 V; close view: 8 nm × 8 nm, 130 pA, 1.5 V; (b) overview (top) and close-up (bottom) after post-annealing at 100 °C. The close-up image shows the co-living of homochiral phase A and racemic phase B. IV conditions: (overview) 260 nm × 260 nm, 80 pA, 1.5 V; close view: 67 pA, 1.5 V (c) after post-annealing at 120 °C. The close-up image shows non-planarized monomers (phase A) with nanographenes (phase C). IV conditions: (overview) 40 nm × 40 nm, 35 pA, 1.5 V. Close view: 15 nm × 15 nm, 84 pA, 1.5 V; (d) after post-annealing at 140 °C. The close-up image shows an island of self-assembled prochiral nanographenes (phase C). IV conditions: overview 90 nm × 90 nm, 40 pA, 1.5 V. Close view: 5 nm × 5 nm, 24 pA, 0.8 V. (e) Schematic representation of the different molecular organizations present throughout the temperature-dependent stages. Molecules have some substituents colored for better clarity: blue six-member rings belong to pyrimidine substituents and red-colored rings indicate upper part of the non-planarized bisanthracene subunits. Dashed red and orange lines indicate N⋯H and Cl⋯H bonding. For the sake of clarity hydrogen atoms are not depicted. | ||
Under annealing at 120 °C (Fig. 2c), the molecular arrangements forming the coordination racemate rows (phase B) are unstable and aggregate into amorphous clusters and short polymers (indicated by white arrows), while the 2D homochiral conglomerates of phase A start to undergo intra-molecular CDH, leading to prochiral, nitrogen-doped nanographenes (phases A + C in Fig. 2c).
It is remarkable to note that this planarization is preferentially promoted in 2D homochiral conglomerate domains of phase A, and the intramolecular transformation preferentially happens in cascade along the direction in which the N⋯H bond is formed, hereafter referred to as the chain direction (Fig. 3 and also sketch of Fig. 2c). The planarization of a single molecule seems to reduce the barrier for the CDH of adjacent molecules (green arrow in Fig. 3) within the same chain as compared to adjacent chains (red arrow in Fig. 3). Interchain N⋯Br interactions could further stabilize the mixed phase, although they might be largely inhibited by the high directionality of the sigma hole of the halogen,36 and the lack of coplanarity between the Br and the N of the pyrimidine in the neighbouring molecule.
 | ||
| Fig. 3 Cascade-effect CDH. STM images of the overview (left) and close-up (right) of the molecular species after a post-annealing to 120 °C. The left image shows both homo- and hetero-chiral moieties, whereas the right image shows details of the homochiral phase. STM IV conditions: (left) 40 nm × 40 nm, 35 pA, 1500 mV; (right) 15 nm × 15 nm, 24 pA, 1500 mV. | ||
At 140 °C the formation of planar and prochiral nanographenes is completed (Fig. 2d). Chiral interactions dominated by intrachain N⋯H and interchain Cl⋯H pairing (dashed red and orange lines respectively in Fig. 2e) lead to the transfer of molecular chirality to the self-assembly, resulting in a racemic distribution of homochiral assemblies of nanographenes (see also Fig. S4).
The significant temperature reduction of the CDH step, which is drastically decreased from 300 °C in route 1 to 140 °C in route 2.2, is tentatively attributed to three interconnected factors: (i) the presence of chlorine atoms in the molecular structure, whose role in assisting CDH is also evidenced in other DBBA derivatives,22,33 (ii) steric hindrance and (iii) cascade planarization.37 In addition, the molecular islands undergo a significant reduction in size, having their periphery decorated with short, randomly dispersed polymers, which may again be an indication that releasing molecular degrees of freedom by diffusion onto the surface facilitates the Ullmann coupling reaction. Viewed the other way round, the limitation of degrees of freedom imposed by molecular self-assembly inhibits Ullmann coupling within the islands until molecules planarize completely at 140 °C forming prochiral islands. From the homochiral character of the islands and the observed size reduction, we suggest that only phase A transforms into planar nanographenes. This is in line with the observation of phase B always at the periphery of the islands, where it can diffuse out and form polymers or other irregular chains. The STM images suggest (confirmed by the XPS analysis described below) that at this stage the bromine atoms are missing at the meso- position of the zig–zag edges. We therefore expect that the C sigma radicals are passivated by the hydrogen released from the CDH reaction. The absence of Br adatoms is also attributed to the hydrogenative desorption as HBr.38,39 Importantly, the chlorine atoms are still present in the molecule, as suggested by the pronounced prochiral appearance of the nanographenes in high resolution STM images (close-up image of Fig. 2d), and confirmed by the XPS data (see below).
In a final step, the post-annealing temperature was slightly increased by 10 °C up to 150 °C (see SI Fig. S5), which promoted the covalent interconnection of nanographenes into 1D chains, most probably through C–Cl bond activation and subsequent Ullmann coupling. Remarkably, the Ullmann polymerization takes place 150 °C below the interribbon coupling that leads to N-NPG using the same precursor, achieved by either the same dechlorinative homocoupling28 or the H–Cl zipping33 reactions. Yet, some non-selective coupling seems to occur, based on the meandering, interconnected morphology of the chains. The whole reaction route is schematically illustrated in Fig. S6.
In order to shed light on the chemical composition of the intermediates and final products, we performed XPS after each of the annealing steps (Fig. 4), tracking C 1s, N 1s, Br 3d and Cl 2p XPS core level spectra and comparing routes 1 and 2.2 that lead to N-NPG and the nanographenes respectively. Our analysis focuses on the halogens, with the aim of tracking the reaction steps in which they are involved (the full set of spectra can be found in Fig. S7). At room temperature and high coverage deposition (route 2.2, bottom panel in Fig. 4a) both spin–orbit coupling Br 3d and Cl 2p doublets are found: Br 3d3/2 and 3d5/2 at 70.9 eV and 69.9 eV respectively31 and Cl 2p1/2 and 2p3/2 at 201.8 eV and 200.2 eV respectively.40 At 100 °C both routes show a new Br 3d doublet at lower binding energies (67.8 eV and 68.8 eV), corresponding to the bromine atoms bonded to the gold surface, an indication of the onset of debromination. Besides, no dechlorination takes place in any of the routes, as no shift is evidenced. After annealing at 140 °C all Br 3d components vanish for route 2.2, in contrast to reaction path 1 where the Br 3d signal is still observable. As previously reported,38,39,41 we correlate the absence of the Br–Au signal to the hydrogenative desorption of Br, which is in turn a fingerprint of the onset of the CDH reactions that planarize the monomers. On the other hand, regarding the Cl 2p doublet, we observe a decrease to 40% of the total raw area along with a shift to higher energies. This decrease in route 2.2 as compared to 1 may be attributed to the early polymerization of nanographenes. The lack of a Cl–Au component indicates that the cleaved Cl directly desorbs, as expected because residual hydrogen is still available according to stoichiometry (CDH releases 8 hydrogen atoms per molecule, and only 4 are used for HBr desorption and passivation of debrominated C sites). At this point of the reaction, the Cl 2p doublet undergoes a shift of 0.5 eV to higher binding energies (Fig. 4a). We consider this shift, not observed during route 1, as a fingerprint of planarization, which results in an increase of the Cl–Au distance and the consequent reduction of charge transfer or screening by Au electrons. Finally, at 200 °C the remaining Cl signal totally vanishes in route 2.2 (Fig. 4a), meaning that all molecules are dechlorinated and Cl has desorbed from the surface. This is in contrast to the prevalence of more than 60% of the original Cl signal for route 1 (Fig. 4b), and agrees with the 1D nanographene chains observed in the STM images above 150 °C. In conclusion, a comparison of the thermal evolution of the core-levels of both routes clearly reveals the different reaction steps followed in each route, and the reduction of the CDH step in the formation of nanographenes.
 | ||
| Fig. 4 XPS characterization of monomer M following different routes, by checking the intensity of the core level XPS peaks as a function of binding energy (BE). (a) and (b) Br 3d and Cl 2p core level XPS spectra of the monomer M after sequential post-annealing temperatures, following route 2.2 (a) or 1 (b). Core level XPS characteristics are displayed in Tables S1 and S2. | ||
Conclusions
We have demonstrated a synthetic route for the on-surface synthesis of a well-defined prochiral, chloro-substituted nitrogen-doped ovalene derivative on Au(111) assisted by the steric hindrance resulting from the molecular self-assembly. Collectively, the presence of the functional group and the steric hindrance originated from the self-assembly reduce the CDH step that leads to planar nanographenes from 300–140 °C. Saturation of the monolayer within a terrace is necessary to block molecular rearrangement and inhibit the Ullmann polymerization that drives the route towards the formation of GNRs. The route was tracked via STM and XPS at different steps of post-annealing, and compared with the formation of the nitrogen-functionalized nanoporous graphene achieved when deposition of the substrate was conducted at the Ullmann polymerization temperature. In STM, homochiral molecular islands at RT turn into two coexisting phases after annealing to 100 °C: the homochiral phase formed by pristine monomers and a new racemic molecular organization presumably induced by coordination to Au adatoms. We have identified the early onset of cyclodehydrogenation at 120 °C exclusively in the homochiral domains, completed at 140 °C. This can be tracked directly from the appearance change of the monomer units in STM images, and indirectly from the hydrogenative desorption of halogens identified by XPS. The observed temperature reduction is essential for the stability of molecules that bear heteroatoms, functional groups or other reactive sites. The new, steric route presented here to synthesize nanographenes at very mild reaction temperatures is expected to be extended to the on-surface synthesis of other heteroatom-doped nanographene compounds.Author contributions
MT: data curation, formal analysis, investigation, writing original draft, review and editing; CM: conceptualization, data curation, investigation, formal analysis, funding acquisition, methodology, project administration, resources, supervision, validation, review and editing; JC: data curation, formal analysis, investigation, review and editing; MVV: data curation, formal analysis, investigation, review and editing; DP: supervision, validation, writing review, editing, funding acquisition, project administration and resources; AM: conceptualization, funding acquisition, project administration, resources, supervision, validation, writing original draft, review and editing.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc07197f.
Acknowledgements
The authors acknowledge Guillaume Sauthier for assisting us with the XPS experiments. This research was funded by the CERCA Programme/Generalitat de Catalunya and supported by Grant No. CEX2021-001214-S, PID2022-140845OB-C63, PID2022-140845OB-C62, and PGC2018-096955-B-C43 funded by MCIN/AEI/10.13039/501100011033, Grant No. TED2021-132388B-C41 and TED2021-132388B-C42 funded by MCIN/AEI/10.13039/501100011033 and the European Union NextGenerationEU/PRTR, the GenCat (Grant No. 2017SGR1506), Xunta de Galicia (Centro de Investigación do Sistema Universitario de Galicia, 2023–2027, ED431G 2023/03) and the European Union (European Regional Development Fund_ERDF). CM was supported by Grant RYC2019-028110-I funded by MICIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”, SUBVTC-2022-0010, PID2022-140827OB-I00 funded by MICIU/AEI/10.13039/501100011033 and by FEDER, UE, and CNS2023-144257 funded by MICIU/AEI/10.13039/501100011033 and by NextGenerationEU/PRTR. M. T. was supported by the Spanish State Research Agency/FSE (ref. BES-2017-08078; project ref. SEV-2013-0295-17-2). DP acknowledges funding from the European Union through the ERC Synergy Grant MolDAM (951519).Notes and references
- V. Bonal, R. Muñoz-Mármol, F. Gordillo Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 3327 CrossRef PubMed.
- Q. Chen, S. Thoms, S. Stöttinger, D. Schollmeyer, K. Müllen, A. Narita and T. Basché, J. Am. Chem. Soc., 2019, 141, 16439–16449 CrossRef CAS PubMed.
- G. M. Paternò, Goudappagouda, Q. Chen, G. Lanzani, F. Scotognella and A. Narita, Adv. Opt. Mater., 2021, 9, 2100508 CrossRef.
- R. Yu, J. D. Cox and F. J. G. de Abajo, Phys. Rev. Lett., 2016, 117, 123904 CrossRef PubMed.
- K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed.
- Z. Liu, S. Fu, X. Liu, A. Narita, P. Samorì, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, 2106055 CrossRef PubMed.
- P. Fantuzzi, A. Candini, Q. Chen, X. Yao, T. Dumslaff, N. Mishra, C. Coletti, K. Müllen, A. Narita and M. Affronte, J. Phys. Chem. C, 2019, 123, 26490–26497 CrossRef CAS.
- Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499–11524 CrossRef CAS PubMed.
- Q. Zhong, B. Viktor, C. Dániel, K. Niu, G. Marcin, A. Ghosh, B. Jonas, E. Daniel, C. Lifeng, S. André and M. C. Stuparu, CCS Chem., 2023, 5, 2888–2896 CrossRef CAS.
- A. Shiotari, K. Tanaka, T. Nakae, S. Mori, T. Okujima, H. Uno, H. Sakaguchi and Y. Sugimoto, J. Phys. Chem. C, 2018, 122, 4997–5003 CrossRef CAS.
- K.-H. Ernst, Acc. Chem. Res., 2016, 49, 1182–1190 CrossRef CAS PubMed.
- E. Turco, S. Mishra, J. Melidonie, K. Eimre, S. Obermann, C. A. Pignedoli, R. Fasel, X. Feng and P. Ruffieux, J. Phys. Chem. Lett., 2021, 12, 8314–8319 CrossRef CAS PubMed.
- S. Mishra, G. Catarina, F. Wu, R. Ortiz, D. Jacob, K. Eimre, J. Ma, C. A. Pignedoli, X. Feng, P. Ruffieux, J. Fernández-Rossier and R. Fasel, Nature, 2021, 598, 287–292 CrossRef CAS PubMed.
- S. Mishra, J. Melidonie, K. Eimre, S. Obermann, O. Gröning, C. A. Pignedoli, P. Ruffieux, X. Feng and R. Fasel, Chem. Commun., 2020, 56, 7467–7470 RSC.
- A. Sánchez-Grande, J. I. Urgel, L. Veis, S. Edalatmanesh, J. Santos, K. Lauwaet, P. Mutombo, J. M. Gallego, J. Brabec, P. Beran, D. Nachtigallová, R. Miranda, N. Martín, P. Jelínek and D. Écija, J. Phys. Chem. Lett., 2021, 12, 330–336 CrossRef PubMed.
- S. Mishra, D. Beyer, R. Berger, J. Liu, O. Gröning, J. I. Urgel, K. Müllen, P. Ruffieux, X. Feng and R. Fasel, J. Am. Chem. Soc., 2020, 142, 1147–1152 CrossRef CAS.
- E. Turco, A. Bernhardt, N. Krane, L. Valenta, R. Fasel, M. Juríček and P. Ruffieux, JACS Au, 2023, 3, 1358–1364 CrossRef CAS PubMed.
- M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2011, 3, 61–67 CrossRef CAS.
- P. H. Jacobse, A. van den Hoogenband, M.-E. Moret, R. J. M. Klein Gebbink and I. Swart, Angew. Chem., Int. Ed., 2016, 55, 13052–13055 CrossRef CAS.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- M. Ohtomo, H. Hayashi, K. Hayashi, H. Jippo, J. Zhu, R. Hayashi, J. Yamaguchi, M. Ohfuchi, H. Yamada and S. Sato, ChemPhysChem, 2019, 20, 3366–3372 CrossRef CAS.
- M. Tenorio, C. Moreno, M. Vilas-Varela, J. Castro-Esteban, P. Febrer, M. Pruneda, D. Peña and A. Mugarza, Small Methods, 2024, 8, 2300768 CrossRef CAS.
- T. Wang, A. Berdonces-Layunta, N. Friedrich, M. Vilas-Varela, J. P. Calupitan, J. I. Pascual, D. Peña, D. Casanova, M. Corso and D. G. de Oteyza, J. Am. Chem. Soc., 2022, 144, 4522–4529 CrossRef CAS PubMed.
- X.-Y. Wang, M. Richter, Y. He, J. Björk, A. Riss, R. Rajesh, M. Garnica, F. Hennersdorf, J. J. Weigand, A. Narita, R. Berger, X. Feng, W. Auwärter, J. V. Barth, C.-A. Palma and K. Müllen, Nat. Commun., 2017, 8, 1948 CrossRef PubMed.
- K. Sun, D. Li, T. Kaihara, S. Minakata, Y. Takeda and S. Kawai, Commun. Chem., 2023, 6, 228 CrossRef CAS PubMed.
- G. Zhu, Y. Jiang, Y. Wang, B.-X. Wang, Y. Zheng, Y. Liu, L.-X. Kang, Z. Li, D. Guan, Y. Li, H. Zheng, C. Liu, J. Jia, T. Lin, P.-N. Liu, D.-Y. Li and S. Wang, J. Am. Chem. Soc., 2023, 145, 7136–7146 CrossRef CAS PubMed.
- S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS.
- Y. Shen, G. Tian, H. Huang, Y. He, Q. Xie, F. Song, Y. Lu, P. Wang and Y. Gao, Langmuir, 2017, 33, 2993–2999 CrossRef CAS PubMed.
- K. A. Simonov, A. V. Generalov, A. S. Vinogradov, G. I. Svirskiy, A. A. Cafolla, C. McGuinness, T. Taketsugu, A. Lyalin, N. Mårtensson and A. B. Preobrajenski, Sci. Rep., 2018, 8, 3506 CrossRef CAS PubMed.
- K. A. Simonov, N. A. Vinogradov, A. S. Vinogradov, A. V. Generalov, E. M. Zagrebina, G. I. Svirskiy, A. A. Cafolla, T. Carpy, J. P. Cunniffe, T. Taketsugu, A. Lyalin, N. Mårtensson and A. B. Preobrajenski, ACS Nano, 2015, 9, 8997–9011 CrossRef CAS.
- C. Moreno, M. Panighel, M. Vilas-Varela, G. Sauthier, M. Tenorio, G. Ceballos, D. Peña and A. Mugarza, Chem. Mater., 2019, 31, 331–341 CrossRef CAS.
- C. Sánchez-Sánchez, T. Dienel, O. Deniz, P. Ruffieux, R. Berger, X. Feng, K. Müllen and R. Fasel, ACS Nano, 2016, 10, 8006–8011 CrossRef.
- M. Tenorio, C. Moreno, P. Febrer, J. Castro-Esteban, P. Ordejón, D. Peña, M. Pruneda and A. Mugarza, Adv. Mater., 2022, 34, 2110099 CrossRef CAS PubMed.
- J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef CAS PubMed.
- J. Björk, C. Sánchez-Sánchez, Q. Chen, C. A. Pignedoli, J. Rosen, P. Ruffieux, X. Feng, A. Narita, K. Müllen and R. Fasel, Angew. Chem., Int. Ed., 2022, 61, e202212354 CrossRef.
- J. Tschakert, Q. Zhong, D. Martin-Jimenez, J. Carracedo-Cosme, C. Romero-Muñiz, P. Henkel, T. Schlöder, S. Ahles, D. Mollenhauer, H. A. Wegner, P. Pou, R. Pérez, A. Schirmeisen and D. Ebeling, Nat. Commun., 2020, 11, 5630 CrossRef CAS PubMed.
- C. Ma, Z. Xiao, H. Zhang, L. Liang, J. Huang, W. Lu, B. G. Sumpter, K. Hong, J. Bernholc and A.-P. Li, Nat. Commun., 2017, 8, 14815 CrossRef CAS.
- C. Bronner, J. Björk and P. Tegeder, J. Phys. Chem. C, 2015, 119, 486–493 CrossRef CAS.
- M. Abyazisani, J. M. MacLeod and J. Lipton-Duffin, ACS Nano, 2019, 13, 9270–9278 CrossRef CAS PubMed.
- M. R. Ajayakumar, C. Moreno, I. Alcón, F. Illas, C. Rovira, J. Veciana, S. T. Bromley, A. Mugarza and M. Mas-Torrent, J. Phys. Chem. Lett., 2020, 11, 3897–3904 CrossRef CAS PubMed.
- A. Mairena, M. Baljozovic, M. Kawecki, K. Grenader, M. Wienke, K. Martin, L. Bernard, N. Avarvari, A. Terfort, K.-H. Ernst and C. Wäckerlin, Chem. Sci., 2019, 10, 2998–3004 RSC.
Footnotes |
| † Present address: Department of Materials, ETH Zurich, CH 8093 Zurich, Switzerland. |
| ‡ Present address: Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue Cambridge, MA, 02139, United States. |
| This journal is © The Royal Society of Chemistry 2026 |