DOI:
10.1039/D5SC07123B
(Edge Article)
Chem. Sci., 2026,
17, 2273-2280
Fast-switching pH-responsive biomimetic ion channels with bidirectional gating control
Received
15th September 2025
, Accepted 25th November 2025
First published on 26th November 2025
Abstract
Acid-sensing ion channels (ASICs) achieve millisecond-gated control over ion permeation through global conformational shifts induced by their acidic pockets. Inspired by this mechanism, we developed a pH-responsive artificial transmembrane transport system by biomimetically reconstructing an acidic pocket domain through the incorporation of a carboxyl-rich cluster into a pillararene–cyclodextrin hybrid scaffold. Transmembrane transport experiments confirmed that this artificial system forms stable, cation-selective ion channels, with its carboxyl groups acting as pH sensors to mediate reversible switching between ON and OFF transport states. Crucially, the gating mechanism is driven by pH-triggered in situ conformational changes, mirroring that of natural ASICs. Stopped-flow experiments further demonstrated that this biomimetic system exhibits millisecond-timescale gating kinetics under pH modulation, achieving response rates comparable to those of natural ion channels.
Introduction
Most channel proteins undergo conformational changes triggered by specific stimuli, which is known as gating. In the gating process, the sub-structural units of channel proteins rearrange to either open or close their ion-permeation pathways, enabling precise control over the flow of ions and molecules.1 For example, acid-sensing ion channels (ASICs) are cation-selective, proton-gated ion channels implicated in pain perception, mechanosensation, and learning and memory processes.2 The gating process of ASICs initiates with the protonation of multiple acidic amino acid residues (Glu 220 and 239 and Asp 238, 346, 350, and 408) situated within the extracellular acidic pockets upon exposure to low pH (Fig. 1). This protonation triggers a collapse of the acidic pockets, which are typically expanded at physiological pH levels.3 Subsequently, the conformational changes resulting from this collapse are relayed through the palm domain to the transmembrane domain, inducing a clockwise rotation of the tilted transmembrane helices and leading to the expansion of the channel gate defined by Glu 426 and Asp 433. This results in an iris-like opening of the ion permeation pathway, enabling ions to pass through the channel. Upon return to physiological pH values, the electrostatic repulsion arising from the deprotonation of acidic residues drives the expansion of the acidic pockets, thereby allowing the transmembrane domain to revert to a non-channel conformation.4 When a variation in extracellular pH occurs, ASICs swiftly transition their ion permeation pathway between the open and closed states within milliseconds, thereby ensuring a timely response to external stimuli.5
 |
| | Fig. 1 Structural features of acid-sensing ion channel 1 (ASIC1) (PDB ID: 2QTS). The transmembrane domain is highlighted in the orange box, showing both top and side views. Aspartic acid and glutamic acid residues involved in the gating process are visually represented by yellow and red spherical models, respectively. | |
Stimuli-responsive channel proteins such as ASICs exhibit a seamless synchronization among their sub-structural units, enabling rapid global conformational shifts in response to external stimuli.4,5 Chemists are fascinated by the molecular mechanisms behind this precise synergy and have conducted significant research endeavors to replicate this process in artificial systems,6,7 aiming to achieve spatiotemporal control over transmembrane transport. A variety of artificial systems that respond to a range of stimuli including pH,8 light,9 voltage,8g,10 ligands,11 mechanical signals and redox stimuli have been explored.12,13 These studies not only offer pioneering approaches for the development of stimulus-responsive artificial transmembrane transport systems, but also offer inspiring insights into their applications in medicine and nanotechnology.14 However, stimulus-responsive artificial systems that can replicate the precise and reversible switching between active and inactive states observed in natural channel proteins remain rare.11c,14h,15 The limited progress in this area poses a significant challenge to achieving spatiotemporal control over transmembrane transport using artificial systems. Herein, we designed a pH-responsive artificial transmembrane transport channel through biomimetic reconstruction of the acidic pocket domain from ASICs. This artificial channel achieves pH-controlled global conformational rearrangements via protonation/deprotonation cycles in the engineered acidic pocket, enabling reversible formation and disintegration of the ion-permeation pathway. Moreover, such a bioinspired system demonstrates millisecond-scale reversible switching between “ON” and “OFF” states under external pH modulation, which achieves a response rate comparable to that of natural channel proteins.
Results and discussion
Design and synthesis
As noted, the acidic pocket in ASICs is the key structural domain responsible for pH-gating during transmembrane ion transport. We hypothesized that if the acidic pocket of ASICs could be reconstructed in artificial systems and utilized to regulate the formation and disintegration of ion-permeation pathways through their protonation/deprotonation cycles, a new class of pH-responsive ion channels could be developed. Thus, we designed a novel class of pillararene–cyclodextrin hybrid molecules by incorporating multiple carboxyl groups at the modifiable sites of the pillararene scaffold (Fig. 2a). Specifically, the carboxyl-rich clusters in these molecules enable us to reconstruct the acidic pocket present in ASICs within artificial systems. These carboxyl groups not only bridge the structural gap between pillararene and cyclodextrin moieties, enhancing the integrity of ion-permeation pathways, but also have the potential to serve as pH-tunable switches, regulating the formation and disintegration of these pathways (Fig. 2b). The hybrid molecules 1 and 2 were prepared by the click reaction of the corresponding bialkynyl-pillar[5]arene and mono-6-azide-6-deoxy-α-cyclodextrin (α-CD-N3), and the structures were characterized using nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (see Section 2 in the SI).
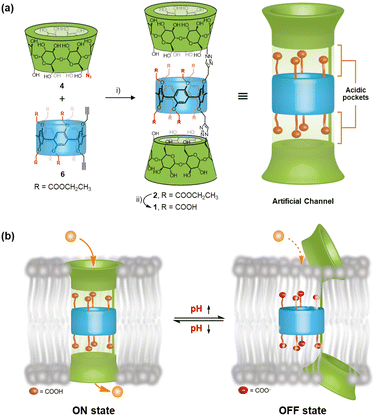 |
| | Fig. 2 (a) Synthesis and structural features of channels 1 and 2: (i) CuI, DIPEA, dry THF; (ii) NaOH(aq.), and THF. (b) Schematic representation of carboxyl groups as a pH-sensing switch that regulates the ON and OFF transport states. | |
Ion transport studies
Following the synthesis and characterization of hybrid molecules 1 and 2, we initially evaluated their lipid bilayer incorporation efficiency and transmembrane ion transport capabilities through 8-hydroxy-pyrene-1,3,6-trisulfonic acid trisodium salt (HPTS) assays.16 Briefly, a suspension of large unilamellar vesicles (LUVs) composed of egg yolk L-α-phosphatidylcholine (EYPC) entrapping the pH-sensitive dye 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) was prepared in 10 mM HEPES buffer containing 100 mM NaCl (pH 7.0). Then a pH gradient across the membranes was introduced by the addition of the LUV suspension to a buffer (10 mM HEPES, 100 mM NaCl, and pH 6.0) (Fig. 3a). The ionophoric activities of hybrid molecules 1 and 2 were evaluated by measuring time-dependent changes in HPTS fluorescence intensity following their incorporation into the vesicle suspension. A significant change in HPTS fluorescence intensity was observed for both compounds 1 and 2, indicating that these hybrid molecules could be inserted into the lipid bilayer and mediate transmembrane ion transport (Fig. S19). Through the Hill analysis of the dose–response curves, the EC50 values (representing the effective concentrations required to achieve 50% activity) were determined to be 0.084 ± 0.011 mM and 0.010 ± 0.007 mM for compounds 1 and 2, respectively (Fig. 3b and c). Compound 2 demonstrated a lower EC50 compared to 1, indicating better efficacy in transmembrane ion transport. This result may be attributed to the distinct functional group modifications on the pillararene scaffold, which is the only structural difference between compounds 1 and 2. The carboxylic ester functional groups in compound 2 may provide a suitable hydrophobicity, thereby enhancing its membrane-incorporation capability and facilitating ion transport across lipid bilayers. To investigate the contribution of membrane uptake to the ionophoric activities of molecules 1 and 2, we performed HPTS assays using LUVs in which the molecules were pre-incorporated during preparation. As demonstrated in Fig. S20, the EC50 values for both molecules under preloading conditions were significantly lower (0.006 ± 0.002 mM for 1 and 0.005 ± 0.002 mM for 2), confirming that membrane uptake substantially impacts their transport activity. The Hill coefficients (n ≈ 1) for both molecules indicate that these tubular molecules mediated ion transport in a single-molecular manner.9c,17
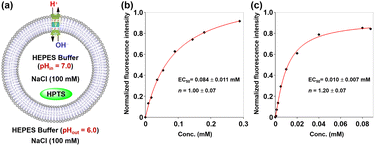 |
| | Fig. 3 (a) Schematic representation of the 8-hydroxypyrene-1,3,6-trisulfonate (HPTS) assay. Changes in normalized fluorescence intensity of HPTS (λex = 460 nm and λem = 510 nm) in vesicles with the concentrations of 1 (b) and 2 (c). | |
To further determine whether the transport activity of the hybrid molecules observed in the above experiments resulted from their disruption of the membrane integrity of LUVs, we performed a carboxyfluorescein (CF) dye leakage assay.13c Briefly, LUVs encapsulating 50 mM carboxyfluorescein (CF), 10 mM HEPES, and 100 mM NaCl (pH 7.5) were prepared, with the external buffer matching the ionic composition and pH while excluding CF. Due to the high intravesicular concentration, CF exists predominantly in a self-quenched dimeric state. Membrane disruption would trigger dye leakage and a consequent fluorescence recovery due to dimer dissociation (Fig. 4a). Notably, no significant fluorescence increase was detected for hybrid molecules 1 and 2 even at 0.063 mM (Fig. 4b). In stark contrast, the positive control melittin (0.015–0.15 µM) induced rapid fluorescence recovery (40.3–87.2%). These data exclude membrane lysis as the origin of the observed ion transport activity for these hybrid molecules.
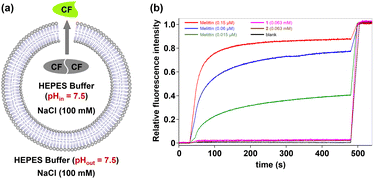 |
| | Fig. 4 (a) Schematic representation of the carboxyfluorescein (CF) dye leakage assay. (b) Changes in the relative fluorescence intensity of CF (λex = 492 nm and λem = 517 nm) in vesicles with time in the presence of 1 and 2 as well as melittin at different concentrations. | |
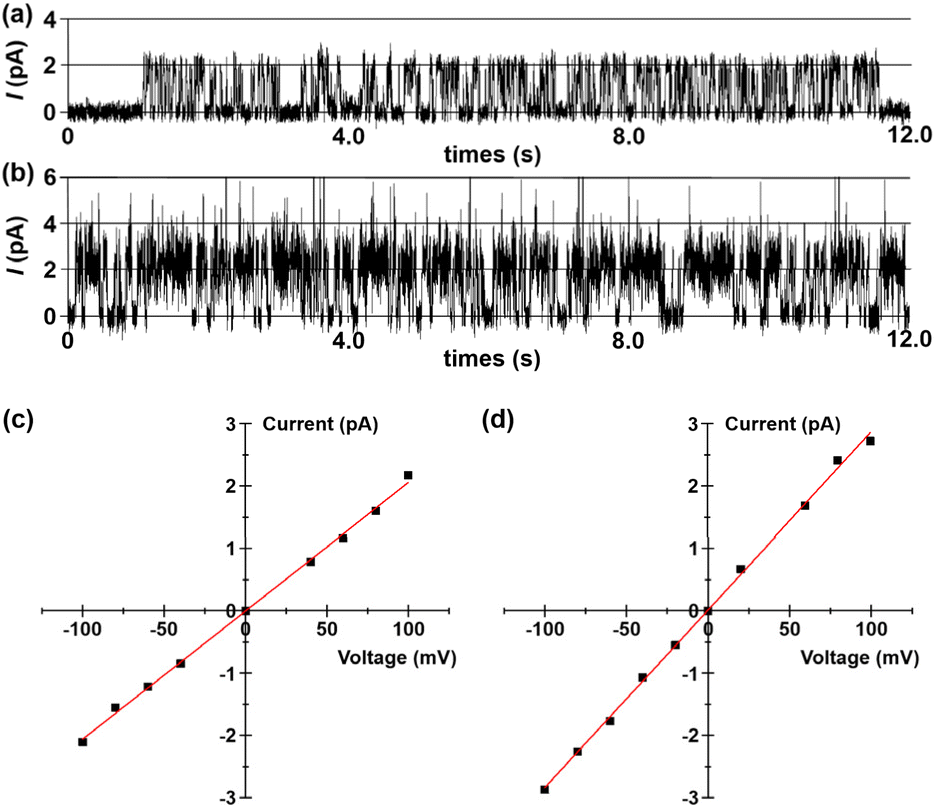
Then, bilayer lipid membrane (BLM) electrophysiology measurements were performed to elucidate the transport mechanism and membrane activity of compounds 1 and 2 (see Section 6 in the SI). For these experiments, a planar lipid bilayer composed of 1,2-diphytanoyl-sn-glycero-3-phosphocholine (diPhyPC) separated two chambers containing 1.0 M KCl solution. Compounds 1 and 2 in DMSO were added to the cis compartment to reach a final concentration of 5 × 10−8 M. Upon applying a voltage of +100 mV across the membrane, regular square-top conductance events were observed for both compounds (Fig. 5a and b). The above observations provide compelling evidence that these hybrid molecules can be incorporated into lipid bilayers to form ion-conducting channels.18 Furthermore, current–voltage (I–V) plots of 1 and 2 were obtained from conductance measurements over a voltage range of −100 to +100 mV. Both compounds exhibited a linear I–V relationship within this voltage range (Fig. 5c and d), with single-channel conductance (γ) values for K+ transport calculated to be 20.6 ± 0.3 pS (1) and 28.5 ± 0.4 pS (2), respectively. The γ value of channel 2 is higher than that of 1, which indicates that channel 2 is more effective in transporting ions.
 |
| | Fig. 5 Current traces through a 1,2-diphytanoyl-sn-glycero-3-phosphocholine (diPhyPC) lipid bilayer in 1.0 M KCl at a potential of +100 mV in the presence of (a) 1 and (b) 2. I–V plots of (c) 1 and (d) 2 in the planar lipid bilayer in a symmetrical 1.0 M KCl. | |
Having established that 1 and 2 mediate the efficient transport of ions through a channel mechanism, BLM electrophysiology measurements in asymmetric bath solutions were further used to probe their ion selectivity. The reversal potentials (Vr) for channels 1 and 2 were determined to be 31.1 mV and 22.3 mV, respectively, by linear fitting of the corresponding I–V plots measured under the condition of cis/trans = 1.0 M/0.5 M KCl bath solutions. The cation/anion permeability ratios PK+/PCl− were determined to be 104.9 (1) and 2.4 (2) using the equation derived from the Goldman–Hodgkin–Katz equation,19 which clearly indicate that these channels possess high cation selectivities (see Section 6 in the SI). Interestingly, channel 1 shows the highest PK+/PCl− value reported for artificial transmembrane channels, far surpassing 2. To elucidate the discrimination mechanism between K+ and Cl− in these ion channels, we performed molecular dynamics simulations (MDS) and determined the energy barriers along the ion permeation pathways using the potential of mean force (PMF). As shown in Fig. S27, for channel 1, the energy barrier for K+ (3.31 kcal mol−1) is significantly lower than that for Cl− (15.64 kcal mol−1). This pronounced difference explains the high K+ selectivity observed in channel 1. Interestingly, Cl− ions exhibit local energy maxima when passing through the carboxyl-rich region, likely resulting from electrostatic repulsion. In contrast, the energy barrier for K+ in channel 2 is only slightly lower than that for Cl− (5.26 vs. 5.71 kcal mol−1), and this narrow margin accounts for its relatively low K+ selectivity.
To further confirm whether channel 1 lacks anion transport capability, fluorescence assays were conducted using a chloride-sensitive 6-methoxy-N-(3-sulfopropyl)quinolinium (SPQ) probe.20 As shown in Fig. 6, when a Cl− concentration gradient was established across LUV membranes, the addition of channel 1 only triggered an 8% rise in SPQ fluorescence intensity compared to the background signal observed in the blank control. This marginal change further demonstrates the low Cl− transport efficiency of channel 1.
 |
| | Fig. 6 (a) Schematic representation of the 6-methoxy-N-(3-sulfopropyl)quinolinium (SPQ) assay. (b) Changes in the relative fluorescence intensity of SPQ (λex = 380 nm and λem = 440 nm) after the addition of 1. | |
Stimuli-responsive ion transport activity
Having elucidated the ion transport mechanisms and behaviours of these hybrid molecules, we next investigated their potential pH-dependent transport activity. As previously mentioned, the ion-permeation pathway of channel 1 contains a carboxyl-rich cluster structurally analogous to the ASIC1 acidic pocket, where pH-sensitive groups may serve as tunable elements to regulate transmembrane transport processes. To test and verify this possibility, the pH-dependent transport activities of channel 1 were evaluated by using HPTS assays at different pH values.8c,d Specifically, LUVs preloaded with an internal buffer containing 10 mM HEPES, 100 mM NaCl (pH 7.0), and 0.1 mM HPTS fluorescent probe were prepared. The LUV suspension was subsequently injected into external buffer solutions (10 mM HEPES and 100 mM NaCl) with varied pH values (4.0, 5.0, 6.0, 8.0, and 9.0) to establish pH gradients across lipid bilayers (Fig. 7a). Following the addition of 1, the fluorescence intensity of HPTS was recorded continuously over a 300-second period. As demonstrated in Fig. 7b, channel 1 exhibited negligible ion transport activity (<1.5%) under alkaline conditions (pH 8.0–9.0). Progressive acidification of the extracellular pH triggered gradual activation of the transport function, with the fluorescence intensity of HPTS increasing from 32% at pH 6.0 to 58% at pH 5.0 and ultimately reaching 64% at pH 4.0. These observations indicate that the transmembrane transport activity of channel 1 demonstrates substantial pH-dependent characteristics. In striking contrast, channel 2, in which the carboxyl groups were substituted with carboxylate ester groups, exhibited pH-independent transport behavior under identical experimental conditions, maintaining stable activity levels between 51% and 61% across the full tested pH range (Fig. 7c). The divergent transport behaviors of these artificial channels within the same pH range imply that the carboxyl-rich cluster in channel 1, which shares the identical scaffold with the pH-insensitive channel 2, may serve as a tunable element to regulate transmembrane transport processes (Fig. 7d).
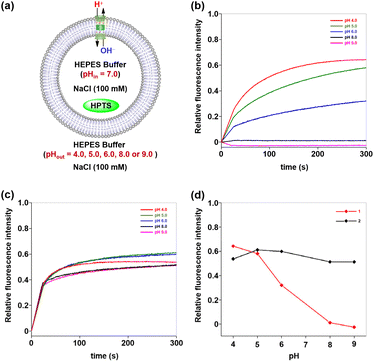 |
| | Fig. 7 (a) Schematic representation of the 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS) assay at different external pH values. Changes in the relative fluorescence intensity of HPTS upon addition of (b) 1 and (c) 2 at different external pH values (* baseline corrected). (d) pH dependence of the transport ability of channels 1 and 2. | |
The high-frequency switching of transmembrane transport processes between ON and OFF states in response to physiological pH variations serves as a hallmark feature of ASICs. The pH-dependent transport behavior exhibited by hybrid molecule 1 under fixed pH gradients has encouraged us to explore whether this molecule could enable ASIC-like rapid switching of transmembrane transport between ON and OFF states in response to extracellular pH changes. To explore this possibility, we developed a real-time, in situ, and pH-tunable HPTS assay that permits direct observation of transport state transitions in artificial channels during pH changes (Fig. 8a). Briefly, LUVs encapsulating 0.1 mM HPTS in 10 mM HEPES buffer (100 mM NaCl, pH 7.0) were prepared and exposed to a transmembrane pH gradient by resuspension in an external buffer (10 mM HEPES, 100 mM NaCl, and pH 8.0). Following the addition of hybrid molecules 1, 2, or gramicidin A (gA) to the LUV suspensions, cyclic pH modulation between 6.0 and 8.0 was achieved through sequential 300-second interval additions of an acid pulse (1 M HCl, 16 µL) or an alkaline pulse (1 M NaOH, 15 µL), with three consecutive cycles monitored via real-time fluorescence spectroscopy. As shown in Fig. 8b, channel 1 demonstrated rapid switching between ON and OFF states in response to extracellular pH changes. When initially exposed to alkaline conditions (pH 8.0) for 300 s, the system exhibited stable fluorescence intensity corresponding to the observed baseline activity (OFF state). Upon acidification of the external buffer to pH 6.0 via an acid pulse, a rapid 96% fluorescence surge indicated that channel 1 switched to an ON state with high ion transport efficiency. Subsequent realkalinization of the external buffer to pH 8.0 triggered a rapid decrease in HPTS relative fluorescence intensity, which likely originates from transient H+/OH− permeation through partially closed channel 1. After 15 s, the fluorescence decay rate stabilized at the baseline level, suggesting that channel 1 had reached a fully OFF state. Notably, channel 1 demonstrated consistent ON/OFF switching efficiency across all three successive pH cycles (6.0 ↔ 8.0), displaying ASIC-like rapid transmembrane gating in response to extracellular pH changes.
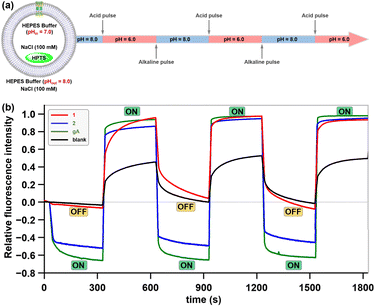 |
| | Fig. 8 (a) Schematic representation of the 8-hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS) assay enabling cyclic modulation of external buffer pH. (b) Time-dependent changes in the relative fluorescence intensity of HPTS upon additions of 1, 2, and gA during cyclic pH modulation (6.0 ↔ 8.0) of the external buffer of LUVs. | |
In contrast, channel 2 (lacking the carboxyl-rich cluster) and the natural cation channel gA exhibited no pH-dependent gating behavior, persistently remaining in an “ON” state throughout three successive pH cycles (6.0 ↔ 8.0) (Fig. 8b). This observation not only provides solid evidence for the key role of the carboxyl-rich cluster in channel 1 during pH-dependent gating, but also validates the designed real-time, in situ HPTS assay as a robust methodology for evaluating pH-dependent gating functionality in artificial transmembrane channels.
Gating mechanism and kinetic characterization
The pH-dependent gating of channel 1 may be governed by two distinct mechanisms: (1) pH-induced protonation/deprotonation of the carboxyl-rich cluster alters its membrane insertion capacity, enabling reversible gating via an insertion–leaving mechanism (Fig. 9a); (2) pH-triggered in situ conformational changes within the lipid bilayer directly alter the ion-permeation pathway, thereby regulating transmembrane ion transport (Fig. 9b). To elucidate the gating mechanism of channel 1, we first employed confocal laser scanning microscopy (CLSM) assay under varying pH conditions to assess the impact of external pH changes on its membrane insertion capacity. For this assay, channel 1 was labeled with 1-oxo-1H-phenalene-2,3-dicarbonitrile (OPD). Then, OPD-labeled channel 1 was incubated with Chinese Hamster Ovary (CHO) cells under pH 5.8 and pH 7.8 for 1 h, respectively. As shown in Fig. 9c and d, CHO cells incubated with OPD-labeled channel 1 exhibited red fluorescence rings at the cell periphery, confirming the successful incorporation of channel 1 into the plasma membrane. However, no significant differences in fluorescence intensity were observed between pH 5.8 and 7.8, suggesting that pH variation within this range does not substantially alter the membrane insertion capacity of channel 1. This finding implies that the insertion–leaving mechanism is unlikely to mediate the reversible switching of this artificial channel between ON and OFF states.
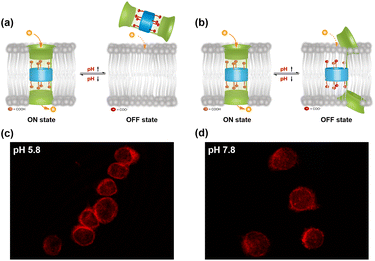 |
| | Fig. 9 Schematic representation of the (a) insertion–leaving mechanism and (b) in situ conformational change mechanism. Confocal laser scanning microscopy (CLSM) images of Chinese Hamster Ovary (CHO) cells after incubation with fluorescently OPD-labeled channel 1 at (c) pH 5.8 and (d) pH 7.8. | |
The natural pH-gated channel proteins ASICs regulate the transmembrane transport process through protonation/deprotonation of acidic residues within their acidic pockets, which triggers a global conformational rearrangement that opens or closes the ion-permeation pathway. Similar to ASICs, channel 1 also contains an acidic pocket formed by a carboxyl-rich cluster. To investigate whether the pH-responsive gating of channel 1 is mediated by conformational changes triggered by protonation/deprotonation of this carboxyl-rich cluster, we performed additional MDS (see Section 10 in the SI). As shown in Fig. 10a, when the carboxyl-rich cluster in channel 1 is in the protonated state, it remains stable within the membrane after 100 ns of simulation, maintaining an intact ion-permeation pathway. Conversely, when the carboxyl-rich cluster is in the deprotonated state, the originally continuous ion-permeation pathway is disrupted due to significant conformational distortion observed after 100 ns of simulation (Fig. 10b). Based on the observations from CLSM and MDS, we can reasonably conclude that the gating mechanism of this molecule is primarily driven by pH-triggered in situ conformational changes, rather than an insertion–leaving process.
 |
| | Fig. 10 (a) Protonated and (b) deprotonated structural models of channel 1 embedded in a 1,2-dioleoyl-sn-glycero-phosphocholine (DOPC) bilayer. | |
MDS studies revealing ultrafast conformational changes in channel molecule 1, along with its rapid gating in pH-tunable HPTS assays (Fig. 8), prompted us to utilize stopped-flow technology to explore the kinetic details of its gating process. Stopped-flow experiments were conducted in two assays: (a) following co-incubation of channel 1 with LUVs (pHin 8.0, pHout 8.0), the suspension was rapidly mixed with an equal volume of acidic buffer (pH 2.74) to achieve a final external pH of 6.0; (b) after co-incubation of channel 1 with LUVs (pHin 6.0, pHout 6.0), the suspension was mixed with alkaline buffer (pH 11.31) to reach the final external pH of 8.0. Stopped-flow technology enabled millisecond-resolution monitoring, revealing a striking divergence in the kinetics of relative fluorescence intensity between the two assays within the initial 150 ms (Fig. 11a). In the first assay (pH 8.0 → 6.0, red line), the relative fluorescence intensity remained stable for the initial 17 ms and then increased rapidly. This rapid rise likely reflects the transition of channel molecules from an OFF to an ON state. In the second assay (pH 6.0 → 8.0, blue line), intensity rose rapidly within the first 38 ms, but its rate of increase slowed significantly thereafter. This may be caused by the channel molecules gradually switching OFF. These distinct millisecond-scale dynamics were reflected in and support the longer-term fluorescence trends observed (Fig. 11b). The first assay exhibited a continuous increase in relative fluorescence intensity after the external pH transition from 8.0 to 6.0, indicating persistent channel opening (ON state), consistent with the rapid activation seen shortly after mixing. In contrast, the second assay showed a rapid increase in fluorescence intensity followed by stabilization after the acidic-to-alkaline shift (pH 6.0 → 8.0), demonstrating an ON-to-OFF transition and stabilization in the OFF state. These results suggest that such molecules may undergo gating state transitions on millisecond timescales, providing experimental evidence for a rapid regulation mechanism in this artificial pH-responsive ion channel.
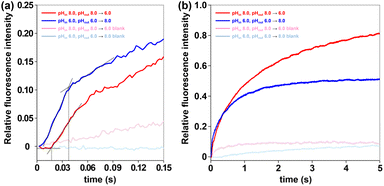 |
| | Fig. 11 HPTS relative fluorescence intensity after pH shift: (a) 0–150 ms; (b) 0–5 s. Vesicles loaded with HPTS (0.1 mM) were pre-incubated with DMSO (control) or 1 (0.1 mM) for 20 min. At t = 0 s, the vesicle solutions were quickly mixed (dead time = 1.2 ms) with the corresponding acidic or alkaline buffer. | |
Conclusions
Inspired by natural pH-gated ion channels like ASICs, which utilize acidic pockets to regulate ion permeation via protonation-induced conformational transitions, we developed a pH-responsive artificial transmembrane transport system by biomimetically reconstructing an acidic pocket domain through the incorporation of a carboxyl-rich cluster into the pillararene–cyclodextrin hybrid scaffold. Electrophysiological studies confirmed that this artificial system formed a stable, cation-selective ion channel, with their carboxyl groups acting as pH sensors to mediate reversible switching between ON and OFF transport states. More importantly, CLSM and MDS studies revealed that the gating mechanism of this molecule is primarily driven by pH-triggered in situ conformational changes, which is consistent with the gating mechanism of natural channel proteins such as ASICs. Furthermore, stopped-flow experiments demonstrated that this biomimetic system exhibited millisecond-timescale gating kinetics under external pH modulation, achieving a response rate comparable to that of natural ion channels. The findings presented herein demonstrate the successful replication of natural channel proteins' gating mechanisms and kinetics in artificial systems – an important step toward systematic biomimicry of natural channels that also holds potential for applications in molecular devices and pharmaceutical technologies.
Author contributions
P. X. and X. C. conceived the idea and designed the research. W. Z. and L. S. synthesized molecules together and performed the ion transport studies and stimuli-responsive ion transport studies. J. C., J. M. and Q. L. finished the gating mechanism and kinetic characterization. N. L. carried out theoretical calculations. P. X. and Y. S. wrote the manuscript of this work. X. C. provided constructive suggestions for results and helped revise the paper. All authors participated in the discussion.
Conflicts of interest
The authors declare no competing interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07123b.
Acknowledgements
We acknowledge funding from the National Natural Science Foundation of China (22271079, 22571073, and 82130103), Research Project from Pingyuan Laboratory (2023PY-ZZ-0201), Central Plains Scholars and Scientists Studio Fund (2018002), Natural Science Foundation of Henan Province (252300420786) and Henan Normal University Research Foundation for the Doctoral Program (QD2023058 and QD2024004). We also acknowledge financial support from the Henan Key Laboratory of Organic Functional Molecules and Drug Innovation.
Notes and references
-
(a)
B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates, Sunderland, 3rd edn, 2001 Search PubMed;
(b)
A. B. Ribera and N. C. Spitzer, Ion Channels, ed. T. Narahashi, Springer, Boston, MA, 1992, vol. 3, pp. 1–38 Search PubMed.
-
(a) E. Lingueglia, J. Biol. Chem., 2007, 282, 17325–17329 CrossRef CAS PubMed;
(b) R. Waldmann, G. Champigny, F. Bassilana, C. Heurteaux and M. Lazdunski, Nature, 1997, 386, 173–177 CrossRef CAS PubMed.
- J. Jasti, H. Furukawa, E. B. Gonzales and E. Gouaux, Nature, 2007, 449, 316–323 CrossRef CAS PubMed.
-
(a) N. Yoder, C. Yoshioka and E. Gouaux, Nature, 2018, 555, 397–401 CrossRef CAS PubMed;
(b) E. B. Gonzales, T. Kawate and E. Gouaux, Nature, 2009, 460, 599–604 CrossRef CAS PubMed.
-
(a) P. Zhang and C. M. Canessa, J. Gen. Physiol., 2002, 120, 553–566 CrossRef CAS;
(b) S. P. Sutherland, C. J. Benson, J. P. Adelman and E. W. McCleskey, Proc. Natl. Acad. Sci. U. S. A., 2000, 98, 711–716 CrossRef PubMed.
-
(a) K. S. Akerfeldt, J. D. Lear, Z. R. Wasserman, L. A. Chung and W. F. DeGrado, Acc. Chem. Res., 1993, 26, 191–197 CrossRef CAS;
(b) X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428–1438 CrossRef CAS PubMed;
(c) A. V. Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef PubMed;
(d) M. Barboiu and A. Gilles, Acc. Chem. Res., 2013, 46, 2814–2823 CrossRef CAS PubMed;
(e) G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824–2833 CrossRef CAS PubMed;
(f) P. Reis and U. Koert, Acc. Chem. Res., 2013, 46, 2773–2780 CrossRef;
(g) M. Mayer and J. Yang, Acc. Chem. Res., 2013, 46, 2998–3008 CrossRef CAS;
(h) T. M. Fyles, Acc. Chem. Res., 2013, 46, 2847–2855 CrossRef CAS PubMed;
(i) B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS;
(j) S. J. Webb, Acc. Chem. Res., 2013, 46, 2878–2887 CrossRef CAS;
(k) F. Otis, M. Auger and N. Voyer, Acc. Chem. Res., 2013, 46, 2934–2943 CrossRef CAS;
(l) J. Montenegro, M. R. Ghadiri and J. R. Granja, Acc. Chem. Res., 2013, 46, 2955–2965 CrossRef CAS PubMed;
(m) Y. Zhao, H. Cho, L. Widanapathirana and S. Zhang, Acc. Chem. Res., 2013, 46, 2763–2772 CrossRef CAS PubMed.
-
(a) S. Chen, Y. Wang, T. Nie, C. Bao, C. Wang, T. Xu, Q. Lin, D.-H. Qu, X. Gong, Y. Yang, L. Zhu and H. Tian, J. Am. Chem. Soc., 2018, 140, 17992–17998 CrossRef CAS;
(b) A. D. Peters, S. Borsley, F. della Sala, D. F. Cairns-Gibson, M. Leonidou, J. Clayden, G. F. S. Whitehead, I. J. Vitórica-Yrezábal, E. Takano, J. Burthem, S. L. Cockroft and S. J. Webb, Chem. Sci., 2020, 11, 7023–7030 RSC;
(c) Y. Ge, W. Li, J. Tian, H. Yu, Z. Wang, M. Wang and Z. Dong, Adv. Sci., 2024, 11, 2400678 CrossRef CAS;
(d) L. Zhang, J. Tian, Z. Lin and Z. Dong, J. Am. Chem. Soc., 2024, 146, 8500–8507 CrossRef CAS PubMed;
(e) Q. Huan, T. Lin, Y.-H. Fu and J.-L. Hou, Chin. Chem. Lett., 2024, 35, 108566–108569 CrossRef CAS;
(f) Q. Xiao, D. Guan, Y.-H. Fu, T. Fan, L. Zhang, Z.-T. Li, Y. Zhang, Y. Wang and J.-L. Hou, J. Am. Chem. Soc., 2024, 146, 22869–22873 CrossRef CAS;
(g) Z. Chen, X. Xie, C. Jia, Q. Zhong, Q. Zhang, D. Luo, Y. Cao, Y. Mu and C. Ren, Angew. Chem., Int. Ed., 2024, 63, e202318811 CrossRef CAS;
(h) Y.-H. Fu, Y.-F. Hu, T. Lin, G.-W. Zhuang, Y.-L. Wang, W.-X. Chen, Z.-T. Li and J.-L. Hou, Nat. Chem., 2024, 16, 1418–1426 CrossRef CAS;
(i) J. Torrisi, M. Chvojka, P. Jurček, X. Zhang, H. Zeng, V. Šindelář and H. Valkenier, Angew. Chem., Int. Ed., 2025, 64, e202424754 CrossRef CAS PubMed;
(j) S. Pophali, D.-D. Su, R. Ata, T. Vijayakanth, S. Nandi, R. Jain, L. J. W. Shimon, R. Misra and M. Barboiu, J. Am. Chem. Soc., 2025, 147, 17404–17415 CrossRef CAS PubMed;
(k) W. G. Ryder, A. Levina, M. E. Graziotto, B. A. Hawkins, D. E. Hibbs, E. J. New and P. A. Gale, Chem, 2025, 11, 102247 CrossRef CAS;
(l) J. Li, O. Catal, I. Marques, D. A. McNaughton, R. M. Maklad, W. G. Ryder, M. J. S. Hill, A. Seddon, W. Lewis, D. J. Adams, V. Félix, X. Wu and P. A. Gale, J. Am. Chem. Soc., 2025, 147, 3392–3401 CrossRef CAS;
(m) S. Chattopadhayay, K. V. Banzal and P. Talukdar, Angew. Chem., Int. Ed., 2025, 64, e202414354 CrossRef CAS;
(n) A. Mondal, D. Mondal, S. Sarkar, U. Shivpuje, J. Mondal and P. Talukdar, Angew. Chem., Int. Ed., 2025, 64, e202415510 CrossRef CAS PubMed;
(o) F. Gou, Q. Wang, Z. Yang, W. Chang, J. Shen and H. Zeng, Angew. Chem., Int. Ed., 2025, 64, e202418304 CrossRef CAS;
(p) Y. Wang, J. L. Hou and W. Wang, Angew. Chem., Int. Ed., 2025, 64, e202502267 CrossRef CAS;
(q) Z. Ye, C. Zhang, Z.-J. Yan, X. Fan, M. Yin, J.-L. Hou and L. Xiao, ACS Nano, 2025, 19, 21057–21067 CrossRef CAS PubMed;
(r) L. Mao, S. Hou, L. Shi, J. Guo, B. Zhu, Y. Sun, J. Chang and P. Xin, Chem. Sci., 2025, 16, 371–377 RSC.
-
(a) G. Das and S. Matile, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5183–5188 CrossRef CAS PubMed;
(b) W.-H. Chen, M. Nishikawa, S.-D. Tan, M. Yamamura, A. Satake and Y. Kobuke, Chem. Commun., 2004, 872–873 RSC;
(c) N. Busschaert, R. B. P. Elmes, D. D. Czech, X. Wu, I. L. Kirby, E. M. Peck, K. D. Hendzel, S. K. Shaw, B. Chan, B. D. Smith, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 3617–3626 RSC;
(d) R. B. P. Elmes, N. Busschaert, D. D. Czech, P. A. Gale and K. A. Jolliffe, Chem. Commun., 2015, 51, 10107–10110 RSC;
(e) E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS;
(f) P. Xin, S. Tan, Y. Wang, Y. Sun, Y. Wang, Y. Xu and C.-P. Chen, Chem. Commun., 2017, 53, 625–628 RSC;
(g) S. P. Zheng, J. J. Jiang, A. van der Lee and M. Barboiu, Angew. Chem., Int. Ed., 2020, 59, 18920–18926 CrossRef CAS PubMed;
(h) T. Yan, S. Liu, C. Li, J. Xu, S. Yu, T. Wang, H. Sun and J. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210214 CrossRef CAS;
(i) W. L. Huang, X. D. Wang, Y. F. Ao, Q. Q. Wang and D. X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302198 CrossRef CAS;
(j) Y. Hu, F. Rigoldi, H. Sun, A. Gautieri and B. Marelli, Chem. Commun., 2023, 59, 10157–10160 RSC;
(k) Y. Wu, Q. Xu, Y. Chen, C. Li, Y. Wu, X. Yu, H. Li, Z. Xu, J. Xu, Z. Ni, Y. Ge, T. Yan, Z. Qi and J. Liu, Adv. Mater., 2025, 37, 2416852 CrossRef CAS PubMed.
-
(a) H. Yang, J. Yi, S. Pang, K. Ye, Z. Ye, Q. Duan, Z. Yan, C. Lian, Y. Yang, L. Zhu, D. H. Qu and C. Bao, Angew. Chem., Int. Ed., 2022, 61, e202204605 CrossRef CAS;
(b) S. A. Gartland, T. G. Johnson, E. Walkley and M. J. Langton, Angew. Chem., Int. Ed., 2023, 62, e202309080 CrossRef CAS PubMed;
(c) Q. Zhong, Y. Cao, X. Xie, Y. Wu, Z. Chen, Q. Zhang, C. Jia, Z. Wu, P. Xin, X. Yan, Z. Zeng and C. Ren, Angew. Chem., Int. Ed., 2023, 63, e202314666 CrossRef;
(d) X. Chao, T. G. Johnson, M.-C. Temian, A. Docker, A. L. D. Wallabregue, A. Scott, S. J. Conway and M. J. Langton, J. Am. Chem. Soc., 2024, 146, 4351–4356 CrossRef CAS;
(e) S. Chattopadhayay, P. Wanjari and P. Talukdar, Chem. Sci., 2024, 15, 17017–17025 RSC;
(f) E. Grählert and M. J. Langton, Angew. Chem., Int. Ed., 2025, 64, e202421580 CrossRef;
(g) C. Li, Y. Wu, S. Bao, H. Li, Z. Xu, J. Yan, X. Yu, L. He, T. Zhang, W. Liu, S. Hou, Y. Zhang, J. Xu, T. Yan, T. Wang, Y. Yan and J. Liu, J. Am. Chem. Soc., 2025, 147, 14139–14153 CrossRef CAS PubMed.
-
(a) T. M. Fyles, D. Loock and X. Zhou, J. Am. Chem. Soc., 1998, 120, 2997–3003 CrossRef CAS;
(b) N. Sakai, D. Gerard and S. Matile, J. Am. Chem. Soc., 2001, 123, 2517–2524 CrossRef CAS PubMed;
(c) W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2014, 53, 4578–4581 CrossRef CAS;
(d) Z.-J. Yan, Y.-W. Li, M. Yang, Y.-H. Fu, R. Wen, W. Wang, Z.-T. Li, Y. Zhang and J.-L. Hou, J. Am. Chem. Soc., 2021, 143, 11332–11336 CrossRef CAS PubMed;
(e) J. F. Lin, X. D. Wang, Y. F. Ao, Q. Q. Wang and D. X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202411702 CrossRef CAS PubMed.
-
(a) P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, Chem.–Eur. J., 2005, 11, 6525–6532 CrossRef CAS;
(b) C. P. Wilson and S. J. Webb, Chem. Commun., 2008, 4007–4009 RSC;
(c) D. Bai, T. Yan, S. Wang, Y. Wang, J. Fu, X. Fang, J. Zhu and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 13602–13607 CrossRef CAS;
(d) R. Sasaki, K. Sato, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2021, 143, 1348–1355 CrossRef CAS PubMed.
-
(a) T. Muraoka, K. Umetsu, K. V. Tabata, T. Hamada, H. Noji, T. Yamashita and K. Kinbara, J. Am. Chem. Soc., 2017, 139, 18016–18023 CrossRef CAS PubMed;
(b) K. Sato, R. Sasaki, R. Matsuda, M. Nakagawa, T. Ekimoto, T. Yamane, M. Ikeguchi, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2022, 144, 11802–11809 CrossRef CAS.
-
(a) J. A. Malla, R. M. Umesh, S. Yousf, S. Mane, S. Sharma, M. Lahiri and P. Talukdar, Angew. Chem., Int. Ed., 2020, 59, 7944–7952 CrossRef CAS;
(b) A. Docker, T. G. Johnson, H. Kuhn, Z. Zhang and M. J. Langton, J. Am. Chem. Soc., 2023, 145, 2661–2668 CrossRef CAS;
(c) L. Shi, W. Zhao, Z. Jiu, J. Guo, Q. Zhu, Y. Sun, B. Zhu, J. Chang and P. Xin, Angew. Chem., Int. Ed., 2024, 63, e202403667 CrossRef CAS PubMed;
(d) C. Li, Y. Wu, S. Bao, Z. Xu, J. Yan, H. Li, X. Yu, Z. Weng, J. Xu, T. Wang, Y. Zhou, T. Yan, Y. Yan and J. Liu, Angew. Chem., Int. Ed., 2025, 64, e202500986 CrossRef CAS.
-
(a) S. Howorka, Nat. Nanotechnol., 2017, 12, 619–630 CrossRef CAS PubMed;
(b) R. D. Mukhopadhyay, Y. Kim, J. Koo and K. Kim, Acc. Chem. Res., 2018, 51, 2730–2738 CrossRef CAS PubMed;
(c) D. P. August, S. Borsley, S. L. Cockroft, F. Sala, D. A. Leigh and S. J. Webb, J. Am. Chem. Soc., 2020, 142, 18859–18865 CrossRef CAS;
(d) M. D Vincenzo, A. Tiraferri, V.-E. Musteata, S. Chisca, R. Sougrat, L.-B. Huang, S. P. Nunes and M. Barboiu, Nat. Nanotechnol., 2020, 16, 190–196 CrossRef PubMed;
(e) Z.-J. Yan, D. Wang, Z. Ye, T. Fan, G. Wu, L. Deng, L. Yang, B. Li, J. Liu, T. Ma, C. Dong, Z.-T. Li, L. Xiao, Y. Wang, W. Wang and J.-L. Hou, J. Am. Chem. Soc., 2020, 142, 15638–15643 CrossRef CAS;
(f) K. Sato, T. Muraoka and K. Kinbara, Acc. Chem. Res., 2021, 54, 3700–3709 CrossRef CAS PubMed;
(g) R. Cao, R. B. Rossdeutcher, Y. Zhong, Y. Shen, D. P. Miller, T. A. Sobiech, X. Wu, L. S. Buitrago, K. Ramcharan, M. I. Gutay, M. F. Figueira, P. Luthra, E. Zurek, T. Szyperski, B. Button, Z. Shao and B. Gong, Nat. Chem., 2023, 15, 1559–1568 CrossRef CAS;
(h) E. Feo and P. A. Gale, Curr. Opin. Chem. Biol., 2024, 83, 102535–102544 CrossRef CAS;
(i) S. Sun, Z. Xu, Z. Lin, W. Chen, Y. Zhang, M. Yan, S. Ren, Q. Liu, H. Zhu, B. Tian, J. Zhang, W. Zhang, S. Jiang, C. Sheng, J. Ge, F. Chen and Z. Dong, Acta Biomater., 2024, 181, 391–401 CrossRef CAS;
(j) A. Kohata and K. Kinbara, Curr. Opin. Chem. Biol., 2025, 84, 102544–102552 CrossRef CAS.
-
(a) M. J. Langton, Nat. Rev. Chem., 2021, 5, 46–61 CrossRef CAS PubMed;
(b) X. Wang, A. Kerckhoffs, J. Riexinger, M. Cornall, M. J. Langton, H. Bayley and Y. Qing, Nat. Nanotechnol., 2025, 20, 432–440 CrossRef CAS PubMed.
- X.-B. Hu, Z. Chen, G. Tang, J.-L. Hou and Z.-T. Li, J. Am. Chem. Soc., 2012, 134, 8384–8387 CrossRef CAS.
-
S. Matile, N. Sakai and A. Hennig, Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley, Chichester, 2012, vol. 8, pp. 473–500 Search PubMed.
-
(a)
R. H. Ashley, Ion Channels: A Practical Approach, Oxford University Press, Oxford, 1995 CrossRef;
(b) J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148–175 RSC.
-
(a) T. M. Fyles, D. Loock, W. F. van Straaten-Nijenhuis and X. Zhou, J. Org. Chem., 1996, 61, 8866–8874 CrossRef CAS;
(b) D. K. Kim, C. H. Duan, Y. F. Chen and A. Majumdar, Microfluid. Nanofluid., 2010, 9, 1215–1224 CrossRef CAS;
(c) J. Feng, M. Graf, K. Liu, D. Ovchinnikov, D. Dumcenco, M. Heiranian, V. Nandigana, N. R. Aluru, A. Kis and A. Radenovic, Nature, 2016, 536, 197–200 CrossRef CAS.
- C. Ren, F. Zeng, J. Shen, F. Chen, A. Roy, S. Zhou, H. Ren and H. Zeng, J. Am. Chem. Soc., 2018, 140, 8817–8826 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Nan
Lv
*a,
Nan
Lv