DOI:
10.1039/D5SC06978E
(Edge Article)
Chem. Sci., 2026,
17, 4168-4173
Efficient singlet fission in rubicene null aggregates
Received
10th September 2025
, Accepted 16th December 2025
First published on 29th December 2025
Abstract
The singlet fission (SF) photovoltaic applications are currently restricted by the limited number of practical SF materials and the lack of understanding of the underlying SF mechanism in typical molecular aggregates. Null-aggregates are molecular aggregates that exhibit minimal exciton–exciton interactions, leading to a monomer-like spectroscopic signature, and thus hold distinct advantages in bypassing extra energy loss and excimer trap issues in conventional H- and J-aggregates. However, it remains unknown if null-aggregates could also contribute to an efficient SF process. In this work, we present an efficient SF system based on rubicene null aggregates. The comprehensive structural and spectroscopic studies demonstrate that the destructive interference between long-range Coulomb and short-range charge-transfer (CT) couplings leads to the monomer-like absorption characteristics of the null aggregates. More importantly, the significant CT coupling interactions contribute to an efficient SF process with a SF rate of (1.0 ps)−1 and a triplet yield of 192% in the null aggregates. Our findings not only provide a deep insight into the SF mechanism in the special null aggregates but also offer a robust SF material system with suitable energies, which would open up a new avenue for the future molecular design and device applications.
Introduction
Singlet fission (SF) that transforms one excited singlet into two triplets has attracted extensive attention owing to its potential in alleviating the thermalization losses and substantially enhancing the conversion efficiency of solar cells.1–7 The range of SF materials continues to grow, and such a MEG process has recently been found in a series of small molecules, oligomers, and conjugated polymers.8–17 Encouragingly, the implementation of the SF-based devices with an external quantum efficiency over 100% represents a huge milestone and has further aroused extensive research interest.5 However, the integration of a highly efficient SF molecular system into real photovoltaic applications remains to be discovered. As a general rule, SF candidates must fulfill the basic energetic requirement that the excitation energy of the singlet state, E(S1), is close to twice that of the triplet state, 2E(T1).1,2 Besides, these materials demand suitable intermolecular interactions to deliver efficient multiexciton generation.1,2 This generally requires the delicate adjusting and engineering of molecular orientation and film morphology in the solid aggregates. Moreover, excessive molecular aggregation might greatly reduce the singlet energy, not only limiting the efficient conversion of high-energy photons but also leading to an over-endothermic SF energetics resulting in the loss of SF. Therefore, aside from basic material design, gaining an in-depth understanding on the underlying SF mechanism in typical molecular aggregates is still a challenging but urgent task.
Molecular exciton theory describes that the staggered “head-to-tail” molecular arrangement forms a J-aggregate exhibiting a red-shifted absorption signature, while the cofacial “side-by-side” packing generates a H-aggregate featuring a blue-shifted absorption characteristic (Scheme 1).18 In recent years, many theoretical and experimental studies have been conducted on the influences of H- and/or J-aggregates on SF dynamics.12,19–27 Nevertheless, the excessive J-type aggregation might greatly reduce the singlet energy leading to not only extra energy loss but also an over-endothermic SF energetics to slow down or even stop the SF process, while the strongly coupled H-type aggregation can usually form an excimer trap state detrimental to the energy conversion process. Null-aggregates, as the name suggests, are molecular aggregates that exhibit minimal exciton–exciton interactions leading to a monomer-like spectroscopic signature in the condensed state.28–32 Thus, such molecular aggregates hold different advantages, which might allow them to overcome the aforementioned extra energy loss and excimer trap issues in the conventional aggregates. Recently, Hong et al. reported efficient multiexciton generation in a dilute solution of a null exciton-coupled perylenediimide folda-dimer.33 However, it remains unknown whether null aggregates could also contribute to an efficient SF process. In this work, we present an efficient SF system based on rubicene (tRc) null aggregates (Scheme 1). The comprehensive structural and spectroscopic measurements suggest that the destructive interference between long-range Coulomb coupling and short-range charge-transfer (CT) coupling leads to the monomer-like absorption characteristics of the current tRc null aggregates. More importantly, strong CT coupling interactions contribute to an efficient SF process with a SF rate, kSF, of (1.0 ± 0.2 ps)−1 and a triplet yield (TY) of 192 ± 30% in the null aggregates. Our findings not only highlight the SF mechanism in the special null aggregates but also offer a robust SF material system, which are important considerations for the future molecular design and device applications.
 |
| | Scheme 1 Efficient SF system based on rubicene (tRc) null aggregates. | |
Results and discussion
Steady state characterization
The polycyclic conjugated hydrocarbon rubicene skeleton, viewed as a C70 fullerene fragment, has recently garnered broad synthetic and optoelectronic application interest.34–40 In the previous work, we employed structural aromaticity to modulate the basic photophysical properties of optoelectronic materials, and designed a new SF material based on the rubicene skeleton with an efficient SF process, and excellent stability.41 In this work, we designed the tert-butyl-substituted rubicene derivative, tRc, in order to further optimize the SF properties and elucidate the underlying SF mechanism in the solid aggregates. The compound was synthesized using the previously reported method (see Section 2 of the SI).
Firstly, we conducted steady-state characterization of tRc molecules in dilute solution and vapor-deposited thin films (Fig. 1). Fig. 1a displays the UV/vis absorption and photoluminescence (PL) spectra. In the monomer state, the molecules show blue-green absorption peaks at 476, 509 and 540 nm, and strong orange emission at 580 nm with a fluorescence lifetime of 4.52 ± 0.01 ns (Fig. 1b). In the thin films, X-ray diffraction (XRD) measurements confirm that they are polycrystalline in nature and exhibit a preferred orientation with the (100) plane parallel to the substrate (Fig. 1c). Fascinatingly, the line shapes of the absorption spectra of the thin films closely match those of monomeric tRc, suggesting null exciton coupling in the condensed state (Fig. 1a).28 The optical band gap (Eg) is estimated to be 2.1 eV, which is determined from the intersection wavelength of the absorption and PL spectra of the thin films. A red-edge absorption characteristic is observed around 600 nm, which could be related to the tail states in the polycrystalline film. These results clearly suggest that tRc films are null aggregates with monomer-like absorption characteristics.
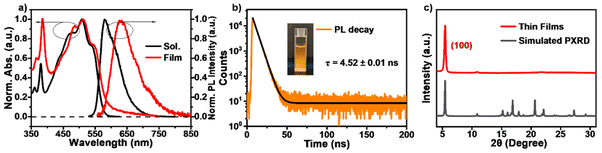 |
| | Fig. 1 Steady state characterization. (a) Normalized UV/vis absorption and PL spectra of tRc molecules in dilute CH2Cl2 solution (10−5 M) and thin films. (b) The PL decay curves of tRc molecules in dilute solution (510 nm excitation). (c) XRD diffractogram of tRc thin films (red) and the simulated powder pattern (black). | |
Subsequently, we investigated the molecular stacking behavior of the studied tRc system in the solid state. The single crystal data display that tRc molecules are arranged in a distinct slip stacking pattern with the π–π distance of 3.42 Å, transverse and longitudinal slip distances of 3.69 and 3.30 Å and slip angles of 46° and 43°, respectively (Fig. 2). To further reveal the nature of the null aggregates, we performed theoretical analysis of exciton coupling interactions in the adjacent molecular dimer unit based on the crystal data of the tRc molecule (for calculation details see Section 4 of the SI).28 The results display that the tRc dimer system exhibits a relative slip angle of 69° between transition dipole moments (Fig. 3). Such an inter-tRc geometry contributes to a positive long-range Coulomb coupling of JCoul = 391 cm−1 and a negative CT-mediated short-range coupling of JCT = −195 cm−1. That is, the destructive interference between Coulomb coupling and CT coupling leads to the monomer-like absorption characteristics in the current tRc null aggregates.28,33 The theoretical results also reflect that the tRc molecule possesses suitable SF energetics given the singlet and triplet excitation energies, E(S1) = 2.30 eV and E(T1) = 1.05 eV, respectively. It should be noted that fluorescence quenching in these null aggregates indicates that an ultrafast nonradiative transition outcompetes and dominates the excited state deactivation process.42–44 These interesting phenomena arouse our curiosity about the excited state photophysics and especially the SF possibility of the current tRc null aggregates.
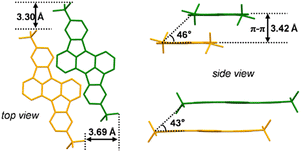 |
| | Fig. 2 Molecular stacking arrangements in tRc crystal structures. | |
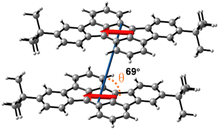 |
| | Fig. 3 The transition dipole moment of the tRc molecule and the relative slip angle between transition dipole moments of adjacent dimer units from the crystal data. | |
Excited state dynamics in dilute solution
Then we applied femtosecond transient absorption (fsTA) measurements to reveal the excited-state deactivation process for the tRc molecule in dilute solution (Fig. 4). As shown in Fig. 4a, the fsTA spectra from dilution solution display negative signals consisting of ground state bleaching (GSB) and stimulated emission (SE) signals in the 480–650 nm range, and the positive excited state absorption (ESA) bands around 650–800 nm. The line shapes of the spectra remain relatively constant over a period of 6.0 ns indicating that no obvious excited state conversion happens and the singlet decay channel dominates the excited state deactivation process. The ESA kinetics at 700 nm decays with two exponentials of τ1 = 148 ± 19 ps and τ2 = 4.0 ± 0.2 ns (Fig. 4b), which are assigned to the minor excited-state structural adjustment and radiative transition processes, respectively. These results clearly demonstrate that the tRc molecule shows typical fluorescence emission behaviour in the monomer excited state.
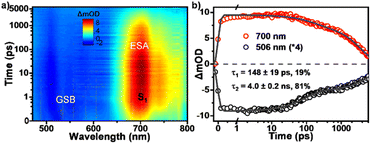 |
| | Fig. 4 Excited state decay dynamics in tRc dilute solution. (a) Pseudocolor plot and (b) kinetic decay curves from fsTA measurements of tRc in dilute solution (450 nm excitation). | |
Robust SF process in null aggregates
We then performed TA measurements on the vapor-deposited films to track the excited state deactivation processes and explore the SF possibility in the studied tRc null aggregates (Fig. 5). The measurements were carried out at a low excitation fluence of 50 µJ cm−2 and the excitation density dependence tests indicated that TA data were free of the heating and annihilation effects of excitation light (Fig. S8). After photo-excitation, the initial fsTA spectra were composed of negative GSB signals around 470–560 nm and positive ESA bands around 560–800 nm, which were assigned to the optically populated S1 state (Fig. 5a and b). Compared to the solution spectral characteristics (Fig. 5a), the TA data show not only a similar ESA signal around 700 nm but also two strong ESA peaks at 750 and 775 nm, which are attributed to the absorption signature of the CT state. Then concurrent with the rapid attenuation of the singlet ESA signals, new ESA bands formed between 470 and 560 nm, strongly overlapping with the GSB signals, and led to substantial cancellation of overall TA signals. The overall changes in the spectral line shapes indicate the formation of new transient species within 80 ps. Then the new species persisted beyond the 6.0 ns detection time window of our fs-TA apparatus. Using parallel samples, nanosecond TA (nsTA) measurements were performed to track the deactivation of the long-lived transient species (Fig. 5c). The TA signals on a time scale of ns-to-µs recovered back to the ground state without further evolution. Given that the line shapes of these signals in the nsTA spectra overlap well with the fs-TA spectral signature, the dominant end species formed upon photo-excitation are undoubtedly individual triplet excitons. The singlet depletion method was then applied to estimate the triplet yield of the tRc films, giving a value of 192 ± 30% (for calculation details see Section 5.4 of the SI).45,46 Given the ultrafast formation rate and high triplet yield, we conclude that an efficient SF process dominates the excited-state deactivation, leading to the observed long-lived triplet populations in the TA spectra. The SF-generated triplet excitons exhibit characteristic biexponential decay kinetics (Fig. S9). These two distinct triplet decay components were then assigned to (i) triplet states undergoing in situ triplet–triplet annihilation back to S0 with τ1 = 219 ± 12 ns (63.2%) and (ii) triplet states that diffuse before annihilation back to S0 with τ2 = 2441 ± 80 ns (36.8%), respectively.41,44 These results suggest that in the current null aggregates, an efficient SF process could still happen to populate long-lived triplet excitons via a CT-mediated mechanism.
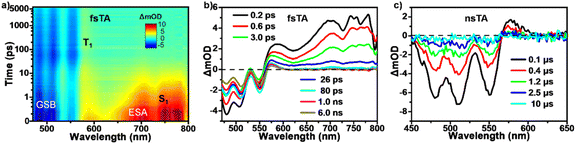 |
| | Fig. 5 SF and triplet decay processes in tRc thin films. (a) Pseudocolor plot and (b) selected spectra from fsTA measurements of tRc thin films (450 nm excitation). (c) nsTA spectra from nanosecond laser flash photolysis measurements of tRc thin films (532 nm-excitation, 50 µJ cm−2). | |
Then we performed global analyses of the fsTA data from tRc thin films (Fig. 6). In order to reflect the polycrystalline characteristics of the thin films, we applied a three-state branched kinetic model (Fig. 6a). The results show that SF proceeds with two rate constants, kSF1 = (1.0 ± 0.2 ps)−1 and kSF2 = (23 ± 4 ps)−1 in the current system (Fig. 6b). The SF timescales of the current molecule seem be closer to those of endothermic materials like tetracenes and other previously reported SF systems.10,14,15 Based on the optical band gap and theoretical triplet energy, the molecule seems to exhibit an iso-energetic SF event. Since theoretical calculations may underestimate the triplet state excitation energy, therefore SF is likely to be an endothermic process in the current tRc system. This further reflects the merit of null-aggregates. They exhibit monomer-like absorption characteristics, which could avoid excessive J-type aggregation that might greatly reduce the singlet energy, not only leading to extra energy loss but also resulting in over-endothermic SF energetics to slow down or even stop the SF process. That is, a population of the optically populated singlet states near hot sites could undergo ultrafast SF with a rate of (1.0 ± 0.2 ps)−1, while the remaining optically excited singlet excitons must diffuse to corresponding hot sites and undergo SF with a slightly slower rate of (23 ± 4 ps)−1 (Scheme 2). Subsequently, these SF-formed long-lived triplet excitons could diffuse and persist over several microseconds in polycrystalline films.
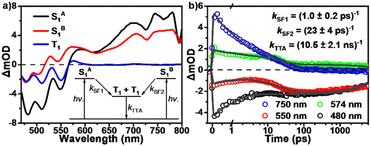 |
| | Fig. 6 Global analysis of the fsTA data of tRc films. (a) Species-associated spectra and kinetic model. (b) Globally fitted wavelength kinetics as well as the corresponding SF and TTA rate constants. | |
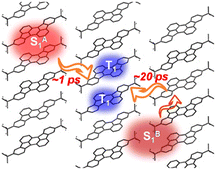 |
| | Scheme 2 Schematic diagram of SF processes in the current tRc system. | |
Discussion
Based on the aforementioned structural and spectroscopic characterization, we conclude that efficient multiexciton generation processes outcompete other singlet decay pathways in the tRc null aggregate system. Compared to conventional H- and J-aggregates, the null aggregate system exhibits monomer-like absorption characteristics. On the one hand, such an aggregate pattern could avoid excessive J-type aggregation that might greatly reduce the singlet energy, not only leading to extra energy loss but also resulting in over-endothermic SF energetics that slow or even stop the SF process. On the other hand, it might also avoid forming an excimer trap state that usually occurs in strongly coupled H-type aggregates. Our results also demonstrate that the destructive interference between long-range Coulomb and short-range CT coupling leads to the monomer-like absorption characteristics of the current null aggregates. Moreover, the results support a CT-mediated SF mechanism in the null aggregate system. More importantly, the current tRc molecule turns out to be a robust SF material system for enhancing the performance of photovoltaic devices given its efficient SF process and suitable energy levels, including E(S1) ∼2.1 eV and E(T1) ∼1.1 eV.1,2
Conclusions
In summary, an efficient SF system based on rubicene null aggregates was successfully fabricated and investigated. Based on comprehensive structural and spectroscopic studies, we demonstrate that the destructive interference between long-range Coulomb and short-range CT coupling leads to the monomer-like absorption characteristics of the current null aggregates. More importantly, strong CT coupling interactions contribute to an efficient SF process with a rate of (1.0 ps)−1 and a triplet yield of 192% in the null aggregates. Our findings not only provide a deep insight into the SF mechanism in the special null aggregates but also offer a robust SF material system with suitable energy levels, which are important considerations for molecular design and device applications.
Author contributions
X. M. S. and L. W. conceptualized and conceived the project. X. M. S. prepared samples and conducted photophysical characterization with the assistance of X. Y. C., Z. Y. L., B. Z., L. P. Y., H. B. F. and L. W. H. Y. and T.-S. Z. performed theoretical calculations. X. M. S. and L. W. wrote the manuscript and all the authors participated in the data analysis and discussions.
Conflicts of interest
There are no conflicts to declare.
Data availability
CCDC 2349726 contains the supplementary crystallographic data for this paper.47
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: molecular synthetic routes and supplementary data for theoretical calculations and TA measurements. See DOI: https://doi.org/10.1039/d5sc06978e.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22501164, 22479107, 22433005 and 22005210) and by the Fundamental Research Program of Shanxi Province (no. 202203021224004 and 20210302124469).
Notes and references
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 Search PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef.
- D. N. Congreve, J. Y. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- C. A. Nelson, N. R. Monahan and X. Y. Zhu, Energy Environ. Sci., 2013, 6, 3508–3519 RSC.
- J. L. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. P. Liu, L. M. Campos and T. L. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, J. Am. Chem. Soc., 2010, 132, 16302–16303 CrossRef CAS PubMed.
- E. Busby, J. L. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- J. H. Hu, K. Xu, L. Shen, Q. Wu, G. Y. He, J. Y. Wang, J. Pei, J. L. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- A. K. Le, J. A. Bender, D. H. Arias, D. E. Cotton, J. C. Johnson and S. T. Roberts, J. Am. Chem. Soc., 2018, 140, 814–826 CrossRef CAS PubMed.
- T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 Search PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 Search PubMed.
- L. Wang, X. Shi, S. Feng, W. Liang, H. Fu and J. Yao, CCS Chem., 2022, 4, 2748–2756 CrossRef CAS.
- S. Wang, X.-Y. Liu, M. Zhang, L. Wang, G. Cui, H. Fu and J. Yao, CCS Chem., 2024, 6, 2142–2149 CrossRef CAS.
- X. Shi, Y. Geng, Z. Wang, E. Zhou, H. Fu and L. Wang, Adv. Funct. Mater., 2025, 35, 2420771 Search PubMed.
- M. Kasha, H. R. Rawls and M. Ashraf El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 Search PubMed.
- A. J. Musser, M. Maiuri, D. Brida, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2015, 137, 5130–5139 CrossRef CAS PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663–671 CrossRef CAS.
- H. Zang, Y. Zhao and W. Liang, J. Phys. Chem. Lett., 2017, 8, 5105–5112 CrossRef CAS PubMed.
- L. Wang, W. Cai, J. Sun, Y. Wu, B. Zhang, X. Tian, S. Guo, W. Liang, H. Fu and J. Yao, J. Phys. Chem. Lett., 2021, 12, 12276–12282 Search PubMed.
- Y. Kim, M. Han, C. Lee and S. Park, J. Phys. Chem. B, 2021, 125, 7967–7974 CrossRef CAS.
- A. Kundu and J. Dasgupta, J. Phys. Chem. Lett., 2021, 12, 1468–1474 CrossRef CAS PubMed.
- A. M. Levine, G. He, G. Bu, P. Ramos, F. Wu, A. Soliman, J. Serrano, D. Pietraru, C. Chan, J. D. Batteas, M. Kowalczyk, S. J. Jang, B. L. Nannenga, M. Y. Sfeir, E. H. R. Tsai and A. B. Braunschweig, J. Phys. Chem. C, 2021, 125, 12207–12213 CrossRef CAS PubMed.
- H. Miyamoto, K. Okada, K. Tokuyama and M. Nakano, J. Phys. Chem. A, 2021, 125, 5585–5600 CrossRef CAS.
- Z. Wu, J. Duan, X. Chen, X. Tian, X. Shi, H. Fu and L. Wang, J. Phys. Chem. Lett., 2025, 16, 9273–9279 CrossRef CAS.
- N. J. Hestand and F. C. Spano, J. Chem. Phys., 2015, 143, 244707 CrossRef.
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS.
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS.
- M. P. Lijina, A. Benny, R. Ramakrishnan, N. G. Nair and M. Hariharan, J. Am. Chem. Soc., 2020, 142, 17393–17402 CrossRef CAS.
- Y. Bo, P. Hou, J. Wan, H. Cao, Y. Liu, L. Xie and D. M. Guldi, Adv. Mater., 2023, 35, 2302664 CrossRef CAS PubMed.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 Search PubMed.
- H. Lee, Y. Zhang, L. Zhang, T. Mirabito, E. K. Burnett, S. Trahan, A. R. Mohebbi, S. C. B. Mannsfeld, F. Wudl and A. L. Briseno, J. Mater. Chem. C, 2014, 2, 3361–3366 RSC.
- X. Gu, X. Xu, H. Li, Z. Liu and Q. Miao, J. Am. Chem. Soc., 2015, 137, 16203–16208 CrossRef CAS PubMed.
- J. Liu, S. Osella, J. Ma, R. Berger, D. Beljonne, D. Schollmeyer, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 8364–8367 CrossRef CAS.
- Y. S. Park, D. J. Dibble, J. Kim, R. C. Lopez, E. Vargas and A. A. Gorodetsky, Angew. Chem., Int. Ed., 2016, 55, 3352–3355 Search PubMed.
- M. Kawamura, E. Tsurumaki and S. Toyota, Synthesis, 2018, 50, 134–138 CrossRef CAS.
- X.-S. Zhang, Y.-Y. Huang, J. Zhang, W. Meng, Q. Peng, R. Kong, Z. Xiao, J. Liu, M. Huang, Y. Yi, L. Chen, Q. Fan, G. Lin, Z. Liu, G. Zhang, L. Jiang and D. Zhang, Angew. Chem., Int. Ed., 2020, 59, 3529–3533 Search PubMed.
- L. Ma, S. Wang, Y. Li, Q. Shi, W. Xie, H. Chen, X. Wang, W. Zhu, L. Jiang, R. Chen, Q. Peng and H. Huang, CCS Chem., 2022, 4, 3669–3676 CrossRef CAS.
- Z. Liu, L. Wang, Q. Deng, J. Zhu, J. Sun, H. Wang, H. Fu and J. Yao, ACS Mater. Lett., 2023, 5, 3010–3016 Search PubMed.
- L. Wang, L. Lin, T.-S. Zhang, S. Guo, Z. Liu, M. Zhang, S. Wang, G. Cui, W.-H. Fang, J. Zhu, H. Fu and J. Yao, CCS Chem., 2023, 5, 2264–2276 CrossRef CAS.
- Z. Wu, C. L. Anderson, T.-S. Zhang, Y. Liu, H. Fu and L. Wang, Chem. Sci., 2025, 16, 13374–13381 RSC.
- Z. Zhao, S. Wang, X. Shi, H. Fu and L. Wang, Chem. Sci., 2025, 16, 5565–5572 RSC.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1–250 CrossRef.
- S. W. Eaton, S. A. Miller, E. A. Margulies, L. E. Shoer, R. D. Schaller and M. R. Wasielewski, J. Phys. Chem. A, 2015, 119, 4151–4161 CrossRef CAS.
-
CCDC 2349726: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2jw2m4.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Lingpeng
Yan
b,
Lingpeng
Yan