
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBead mill-driven acceleration in catalytic methanolysis reaction of poly(ethylene terephthalate) toward low-energy chemical recycling of polymers
Tomoya
Kawase
a,
Haruro
Ishitani
 *b and
Shū
Kobayashi
*ab
*b and
Shū
Kobayashi
*ab
aDepartment of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Binkyo-ku, Tokyo 113-0033, Japan
bGreen & Sustainable Chemistry Social Cooperation Laboratory, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: hishitani@chem.s.u-tokyo.ac.jp; shu_kobayashi@chem.s.u-tokyo.ac.jp
First published on 28th January 2026
Abstract
Chemical recycling of poly(ethylene terephthalate) (PET) is crucial for sustainable resource circulation. However, conventional high-temperature PET depolymerization methods suffer from high energy consumption and efficiency limitations. Here, we demonstrate that a bead milling pretreatment significantly accelerates the methanolysis at temperatures below 90 °C. The effectiveness of this pretreatment is highly dependent on the choice of bead material and solvent, as these factors influence polymer chain scission and molecular weight reduction. Gel permeation chromatography (GPC) analysis revealed that bead milling breaks down PET into lower molecular weight species, and the extent of this reduction is controllable by the solvent's affinity for PET. We also developed a one-pot process that integrates continuous bead milling and methanolysis, achieving PET depolymerization without the need for external heating. We successfully utilized this DMT for the continuous synthesis of pharmaceuticals, highlighting the potential for upcycling commodity polymers. This efficient, low-temperature depolymerization of PET significantly reduces energy consumption compared to conventional high-temperature processes, paving the way for a more environmentally friendly recycling method. This study represents a significant step towards a sustainable society in an era of increasing resource constraints and environmental concerns.
Introduction
Plastics, synthetic polymers, have become indispensable materials in modern life due to their lightness, moldability, and low cost.1,2 Global plastic production has steadily increased and is projected to reach 1 Gt per year within the next 30 years,3–5 unless there is a major shift to alternatives. However, as production increases, the disposal of used plastics remains a persistent issue. Continued reliance on landfilling and incineration is no longer viable.3,6 To address this, scientific and technological advancements must promote a movement toward plastic recycling based on the “reduce, reuse, and recycle” principles.7–9 This challenge aligns with the broader goal of achieving a carbon-circulating society that is not dependent on fossil resources.2,10 Given the current gap in cost-effectiveness between renewable carbon sources (from biomass or captured CO2) and fossil fuels, converting waste resources into high-value-added products is a critical strategy.6,11 In this context, chemical recycling, which involves breaking down polymers into small molecules, offers a promising way to add value to surplus waste plastics.12Many common, mass-produced plastics like poly(ethylene terephthalate) (PET) are solvent-resistant and require thermal melting to promote mass transfer and accelerate chemical reactions.13,14 While thermal energy is unavoidable in mechanical recycling (e.g., remolding), chemical recycling can transform plastics into other materials through reactions that require less energy. Therefore, a key challenge in chemical recycling is overcoming the high temperatures needed for these chemical transformations.2,10,15 Although increasing the surface area of plastics through micronization could enhance reactivity, handling submicron-sized particles from physical pulverization is difficult16–21 due to the thermoplastic nature of polymers.3 The solution lies in the effective dispersion of heat during pulverization and the stabilization of the resulting polymer sub-microparticles.
Based on these considerations, we focused on wet pulverization methods.22–24 This solvent-based approach creates ultrafine particles by leveraging dispersion and comminution effects not present in dry pulverization methods. We hypothesized that by grinding polymers in an appropriate slurry system, excess thermal energy—which would cause polymers to melt—could be dispersed.25–27 Furthermore, we anticipated that the shear and collision energies from the milling media could be used more effectively for micronization, and that solvation by the solvent would stabilize the resulting sub-microparticles. While ball milling is typically associated with dry mechanochemical processes and has recently found increasing use in chemical transformations,28 this study focuses on the unique benefits of the high collision energy achieved in bead milling, which is predominantly operated as a slurry-based wet pulverization system (Fig. 1).29,30 To demonstrate this concept, we investigated the depolymerization of PET into its monomers. While mechanochemical approaches for PET depolymerization have been studied,31–33 our work primarily uses wet pulverization as a pretreatment to prepare substrates for subsequent depolymerization. We systematically evaluated the contribution of this pretreatment to facilitating efficient depolymerization under mild conditions. By elucidating the role of wet pulverization in enhancing chemical reactivity, this study aims to establish a general pretreatment strategy independent of polymer-specific chemical properties, paving the way for low-temperature chemical transformations of a broad range of plastic materials.
 | ||
| Fig. 1 Bead mill treatment. | ||
The most straightforward method for depolymerizing PET into its monomers is alkaline hydrolysis. However, this process requires more than two equivalents of a strong base like sodium hydroxide and inevitably generates large amounts of salt during the neutralization step, which is a significant drawback.34–37 In contrast, alcoholysis offers a catalytic pathway for chemical recycling, primarily aimed at producing the monomer dimethyl terephthalate (DMT).2,13,15,38–44 Although an indirect approach, DMT's high crystallinity facilitates its purification, making it suitable for regenerating high-quality recycled PET or for upcycling into value-added chemicals.41 However, conventional alcoholysis typically requires high temperatures. For example, it is often performed with ethylene glycol at over 180 °C to produce bis(2-hydroxyethyl) terephthalate (BHET),13,15,37,41,44 which is then converted to DMT via homogeneous transesterification.
To address the high-temperature issue, single-step conversion from PET to DMT under milder conditions has been explored, including our preliminary work.45–54 Hirano's group, for instance, recently reported methanolysis using a La(acac)3 catalyst at 150 °C for the direct production of DMT.47 However, high-temperature methanolysis requires high-pressure reaction equipment, which is unsuitable for large-scale processing. Nakajima and Tanaka's group achieved milder conditions by using dimethyl carbonate as a co-solvent to capture the byproduct ethylene glycol and control the equilibrium.51,52 Similarly, Cho's group reported milder conditions using 20 mol% K2CO3 and dichloromethane (DCM) as a co-solvent.53 While these previous studies used co-solvents and equilibrium control to alleviate mass transfer limitations, these approaches often introduce new issues related to cost and environmental impact. Our approach, in contrast, aims to solve these problems by using bead mill treatment as a pretreatment for methanolysis.
Results and discussion
Investigation of bead mill conditions
Given that PET depolymerization is typically performed at temperatures above 180 °C,13,15,39–44,55 we set a key milestone for this study: achieving methanolysis at 90 °C or lower without a co-solvent in depolymerization. To evaluate the wet pulverization strategy, we designed a two-stage process (Fig. 2). First, PET pellets were bead-milled with a solvent to form a slurry. Second, the slurry was dried to a fine PET powder, which was then used for a methanolysis reaction with methanol as the sole solvent and a catalyst in a simple Pyrex-sealed tube. We used commercially available, reagent-grade PET pellets for initial investigations to ensure reproducibility (SI-2-1). We used a 400 mL vertical bead mill with high-speed rotary blades. The standard milling medium was 2 mm diameter zirconia beads, and milling was typically performed at 2500 rpm for 3 hours. The resulting slurry was filtered to remove unpulverized PET and beads, and the solvent was then removed to yield a fine PET powder. | ||
| Fig. 2 Two-step evaluation through bead mill treatment and methanolysis. | ||
In our initial investigations, we performed methanolysis reactions on both bead-milled and untreated samples for 4 hours using tetrabutylammonium hydroxide (TBAOH) as a catalyst (Table 1). Without bead milling, the yield of the target product, DMT, was 14% (Table 1, entry 2). In contrast, bead-milled samples showed a significant acceleration in methanolysis. The versatility of bead milling conditions makes it a promising tool for chemical processing. We found that the choice of solvent was particularly critical. In this preliminary study, testing toluene, water, and acetonitrile as milling solvents, the highest yield (69%) was achieved with toluene (Table 1, entry 12). The bead material was also a key parameter; zirconia beads gave the highest yield, while alumina beads showed minimal effect. While the dynamics of local stress and deformation from bead collisions are complex,56 we believe that the high strength and density of zirconia beads imparted the high collision energy needed for effective milling.57 These initial findings indicated that bead milling effectively converts PET pellets into reactive particles, which significantly enhances methanolysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Bead-milling | Cat.a | Yield [%]b | ||
| Bead | Solv. | DMT | EG | ||
| a W, With catalyst; Wo, without catalyst. b Determined by GC analysis. c PET pellet was utilized directly. d Not detected. e Primary particle size: 27 µm. f Primary particle size: 2.9 µm. g Primary particle size: 4.6 µm. | |||||
| 1c | Wo | Wo | 0 | NDd | |
| 2c | W | 14 | NDd | ||
| 3 | ZrO2 | H2Oe | Wo | 9 | 6 |
| 4 | W | 39 | 28 | ||
| 5 | ZrO2 | MeOH | Wo | 11 | 7 |
| 6 | W | 38 | 26 | ||
| 7 | ZrO2 | CH3CNe | Wo | 14 | 10 |
| 8 | W | 53 | 36 | ||
| 9 | ZrO2 | Benzene | Wo | 28 | 20 |
| 10 | W | 59 | 39 | ||
| 11 | ZrO2 | Toluenef | Wo | 30 | 12 |
| 12 | W | 69 | 42 | ||
| 13 | ZrO2 | CHCl3 | Wo | 23 | 18 |
| 14 | W | 58 | 37 | ||
| 15 | Al2O3 | Tolueneg | Wo | 4 | NDd |
| 16 | W | 16 | 9 | ||
| 17 | Steel | Toluene | Wo | 14 | 11 |
| 18 | W | 46 | 30 | ||
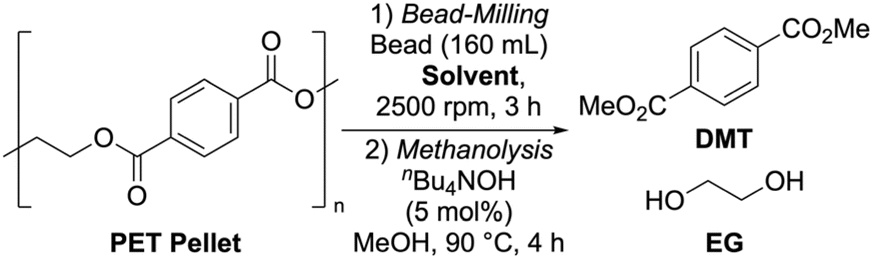
Effect of bead mill treatment on polymer structure
Previous work by Tanaka et al. on methanolysis in dimethyl carbonate compared the reactivity of PET powders in different size classes (53–106 µm, 106–250 µm, and 250–500 µm).51 They found that the smallest fraction had the fastest reaction rate, but the difference in yield was only about 5%. This suggests that a finer particle size does not always lead to a significantly faster reaction rate. To understand the reasons behind the yield differences in our study, we analyzed the particle size distribution of our bead-milled PET samples (SI-3-1).Dynamic light scattering (DLS) measurements revealed that the particles produced gradually aggregated over time. However, the primary particle size obtained from toluene-mediated bead milling was approximately 2.9 µm, which was about one-tenth the size of those obtained with water or acetonitrile, both at 27 µm. These results clearly demonstrate that bead milling significantly reduced the PET particle size, and furthermore that the size was greatly affected by the choice of solvent. Also, the pronounced particle size effect observed in this study contrasts with the prior research, likely due to the finer sub-microparticles being less than 10 µm.
A surprising finding was that alumina beads, despite producing similarly small particles (4.6 µm), resulted in a notably low methanolysis rate. This indicates that factors other than particle size reduction influence methanolysis performance. The high collision energy from zirconia beads likely causes effects beyond simple micronization. We then used gel permeation chromatography (GPC) to analyze the molecular weight of the bead-milled PET samples (Table 2). The untreated PET pellet had a number-average molecular weight (Mn) of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900, a weight-average molecular weight (Mw) of 63700, and a peak-top (Mp) of 54900. After bead milling in toluene, these values were reduced to Mn = 7,380, Mw = 24800, and Mp = 15900. This indicates that the bead milling process cleaves polymer chains, resulting in smaller molecular weight polymers. The minimal change in the polydispersity index (Mw/Mn) suggests a nearly uniform chain scission across the polymer population.
900, a weight-average molecular weight (Mw) of 63700, and a peak-top (Mp) of 54900. After bead milling in toluene, these values were reduced to Mn = 7,380, Mw = 24800, and Mp = 15900. This indicates that the bead milling process cleaves polymer chains, resulting in smaller molecular weight polymers. The minimal change in the polydispersity index (Mw/Mn) suggests a nearly uniform chain scission across the polymer population.
| Entry | Bead-milling | Molecular weight distributionb | Methanolysis resultc | |||||
|---|---|---|---|---|---|---|---|---|
| Bead | Solvent | REDa | M n | M w | M w/Mn | M p | ||
| a Relative energy differences calculated using Hansen's solubility parameters. b Determined by GPC analyses. c Results of methanolysis reaction under the standard conditions. d Without conducting the bead mill treatment. | ||||||||
| 1d | Wo | — | 16900 |
63700 |
3.8 | 54900 |
14 | |
| 2 | ZrO2 | H2O | 4.67 | 13700 |
44500 |
3.3 | 38900 |
39 |
| 3 | ZrO2 | CH3CN | 1.62 | 9620 | 37200 |
3.9 | 33400 |
53 |
| 4 | ZrO2 | Toluene | 0.85 | 7280 | 24800 |
3.4 | 15900 |
69 |
| 5 | ZrO2 | CHCl3 | 0.44 | 15900 |
59000 |
3.7 | 53800 |
58 |
| 6 | Al2O3 | Toluene | 0.85 | 11000 |
41200 |
3.7 | 37800 |
16 |
Two key observations from this analysis were:
(1) The degree of molecular weight reduction correlated with the hierarchy of yields observed among the different solvents. The lower yield from alumina beads, despite producing small particles, might be due to insufficient molecular weight reduction. This suggests that the strong collision energy from zirconia beads was effectively used for PET main chain scission.
(2) The choice of solvent significantly affects both particle size and molecular weight reduction.
The interaction between PET and the solvent appears to be crucial for generating low molecular weight, ultrafine particles. We supported this hypothesis by calculating the Relative Energy Difference (RED) using Hansen Solubility Parameters (HSP), which represents the affinity between the two materials.58–61 Toluene showed the lowest RED value (0.85) among the tested solvents (water, acetonitrile, and toluene), indicating the highest affinity for PET (SI-4). This aligns with the conventional understanding of wet pulverization, where solvents don't necessarily dissolve the material but contribute to particle stabilization through wetting effects. Chloroform, with a significantly lower RED value (0.44) than toluene, can partially dissolve PET. However, bead milling in chloroform caused minimal changes to PET's molecular weight (Table 2, entry 5). We postulate that when PET chains are not fixed in a solvent, the mechanical energy from the bead mill is not effectively transferred to cleave chemical bonds but is instead dispersed as molecular thermal motion. However, methanolysis yields from the chloroform-milled sample were similar to those from the toluene-milled sample. This suggests that the chloroform pretreatment enhanced PET's susceptibility to dissolution, thereby increasing the reactive surface area. Based on these results, we concluded that the accelerated methanolysis reaction is caused by both a reduction in particle size and the cleavage of polymer chains, which leads to the formation of lower molecular weight PET.
Investigation of depolymerization reactions
We further investigated the influence of bead milling conditions on the methanolysis reaction. We found that parameters such as PET pellet dosage, cooling system temperature, and circumferential speed did not significantly affect the methanolysis rate with TBAOH as a catalyst. Interestingly, extending the bead milling time to 15 hours paradoxically decreased the methanolysis yield. Furthermore, no substantial difference in yield was observed when the pulverized PET powder was milled again with smaller (1 mm) beads (Table 3).|
|
|||
|---|---|---|---|
| Entry | Deviation in milling conditions | Yield of DMT [%]b | |
| Wo/TBAOH | W/TBAOH | ||
| a PET pellet purchased from Aldrich (crystallinity, 26%) was used. b Determined by GC analysis. | |||
| 1 | None | 31 | 69 |
| 2 | 10 g of PET was used for the milling | 35 | 65 |
| 3 | 0 °C (cooling) | 30 | 62 |
| 4 | 1000 rpm instead of 2500 rpm | 29 | 60 |
| 5 | 1500 rpm instead of 2500 rpm | 30 | 65 |
| 6 | 6 h instead of 3 h | 32 | 56 |
| 7 | 15 h instead of 3 h | 29 | 52 |
| 8 | Additional milling with Φ 1 mm beads | 29 | 66 |
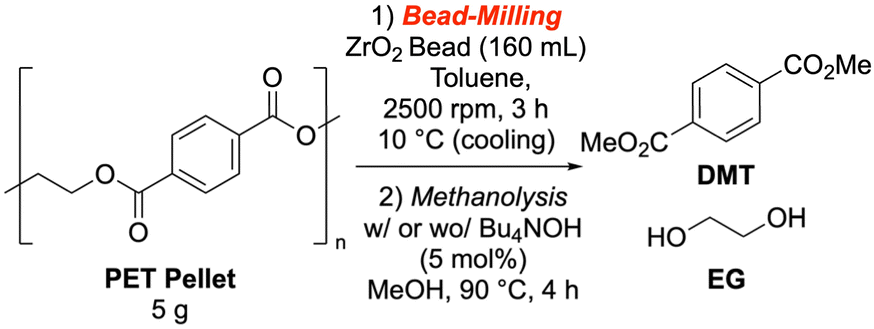
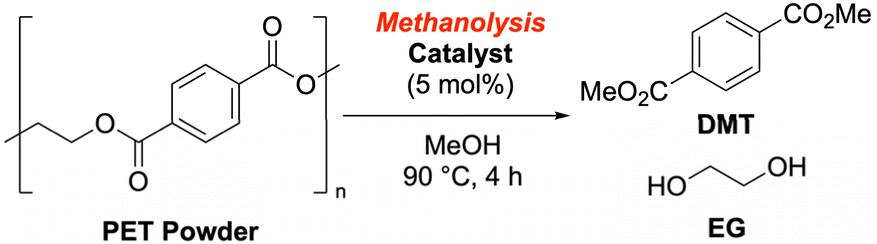
Next, we scrutinized methanolysis reactions using PET powder prepared under standard zirconia bead milling conditions (Table 4). While standard methanolysis was conducted at 90 °C, the reaction at 120 °C proceeded with the catalyst regardless of the milling treatment. However, milling enabled DMT formation at 60 °C, achieving a 37% yield in 4 hours (entry 10), whereas only 1% was obtained without milling (entry 4). Our catalyst screening (Table 5) revealed that potassium methoxide, potassium carbonate, and several quaternary ammonium salts significantly accelerated the reaction (entries 3–6, SI-5-1). Benzyl trimethylammonium hydroxide (entry 9) and tetramethylammonium methoxide (TMAM, entry 10) showed the most remarkable acceleration. With 10 mol% TMAM, DMT was isolated in 81% yield (entry 11). Although we tested the lanthanide Lewis acid conditions reported by Hirano et al.,47 they resulted in relatively low yields under our standard conditions (entry 13).
|
|
||||
|---|---|---|---|---|
| Entry | Bead milling | Catalyst | Temp. [°C] | Yield of DMT [%]b |
| a PET pellet purchased from Aldrich (crystallinity, 26%) was used. b Determined by GC analysis. | ||||
| 1 | Not done | Wo/ | 60 | 0 |
| 2 | Wo/ | 90 | 0 | |
| 3 | Wo/ | 120 | 27 | |
| 4 | W/ | 60 | 1 | |
| 5 | W/ | 90 | 14 | |
| 6 | W/ | 120 | 72 | |
| 7 | Done | Wo/ | 60 | 6 |
| 8 | Wo/ | 90 | 31 | |
| 9 | Wo/ | 120 | 64 | |
| 10 | W/ | 60 | 37 | |
| 11 | W/ | 90 | 69 | |
| 12 | W/ | 120 | 74 | |

|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield [%]a | |
| DMT | EG | ||
| a Determined by GC analysis. b Isolated yield. c The reaction was performed using 10 mol% of the catalyst. d Not detected. | |||
| 1 | Wo/ | 31 | 13 |
| 2 | TBD | 29 | 17 |
| 3 | KOMe | 69 | 44 |
| 4 | NaOMe | 63 | 33 |
| 5 | K2CO3 | 62 | 43 |
| 6 | n Bu4NOH (TBAOH) | 69 | 36 |
| 7 | n Bu4NOAc | 42 | 19 |
| 8 | n Bu4NI | 36 | 16 |
| 9 | PhMe3NOH | 76 | 38 |
| 10 | Me4N(OMe) (TMAM) | 76 | 41 |
| 75b | |||
| 11c | TMAM | 81b | 37 |
| 12 | Me4N(OCO2Me) | 49 | 26 |
| 13 | La(acac)3 | 38 | 17 |
| 14 | La2O3 | 33 | 17 |
| 15 | Zn(OAc)2 | 14 | NDd |
To better understand the reaction profile, we tracked the DMT yield over time in four different reaction systems, varying the presence of bead milling and the TMAM catalyst. For the reagent PET, the bead-milled sample with the TMAM catalyst showed an approximately 60% DMT yield within 30 minutes, compared to less than 1% for the untreated PET (Fig. 3).
 | ||
| Fig. 3 Time course of the methanolysis (1) white squares, using PET pellet and without using catalyst; black squares, using PET pellet in the presence of TMAM catalyst; blue circles, using micronized PET powder and without using catalyst; red circles, using micronized PET powder and in the presence of TMAM catalyst. Yields were determined by GC analyses. | ||
The yield was calculated by correcting for impurities, as the standard reagent PET pellet contained reinforcing agents. To avoid ambiguity, we verified our results using amorphous and crystalline PET pellets (without reinforcers, provided by a supplier) and PET flakes from used beverage bottles. The results consistently showed the significant effect of both bead milling and catalysis across all PET samples (Fig. 4). For the amorphous PET pellet (PET B) and the used beverage PET flakes (PET D), methanolysis with bead milling and TMAM gave a higher DMT yield than with the standard reagent PET pellet (PET A). Reactions without the catalyst or milling showed poor conversions. Similar trends were observed for the crystalline PET pellet (PET C), although with a slightly lower DMT yield (SI-5-2, 5-3).
 | ||
| Fig. 4 Methanolysis results using several PET samples PET A, the standard reagent PET pellet; PET B, the amorphous PET pellet; PET C, the crystalline PET pellet; PET D, the used beverage PET flake: cond. A, using PET pellet and without using catalyst; cond. B, using PET pellet in the presence of TMAM catalyst; cond. C, using micronized PET powder and without using catalyst; cond. D, using micronized PET powder and in the presence of TMAM catalyst. Yields were determined by GC analyses. | ||
We also investigated a gram-scale methanolysis reaction using 1.0 g of bead-milled PET from used beverage PET flakes under standard conditions (Scheme 1). After the reaction, methanol was recovered by evaporation, and the residue was recrystallized with H2O/acetone to yield 0.865 g of analytically pure DMT as white crystals. This demonstrated the scalability and robustness of the process.
 | ||
| Scheme 1 Gram-scale methanolysis and isolation of DMT. | ||
To explore near-room temperature methanolysis, we investigated organic co-solvent systems. Pyridine proved to be the optimal co-solvent, and a methanol/pyridine (1:3) system gave an 85–90% DMT yield within 4 hours at 30 °C using micronized PET from either the amorphous PET pellet or the used beverage PET flakes, while 58% yield was attained for standard reagent PET (Scheme 2, SI-5-4, 5-5).
 | ||
| Scheme 2 Methanolysis under near room temperature conditions using co-solvent system. | ||
This methodology also proved versatile for other solvolysis reactions. Aminolysis with 2-aminoethanol13,38,62,63 using pyridine as the main solvent yielded 79% bis-hydroxyethyl terephthalamide (BHETA) from the reagent PET pellet at 30 °C in 16 hours, and an impressive 96% yield from used beverage PET flakes in just 1 hour. Glycolysis with ethylene glycol13–15,38–41,44,64 proceeded efficiently under mild conditions, producing BHET quantitatively at 60 °C in acetonitrile within 24 hours. These results conclusively demonstrate that bead milling significantly contributes to low-energy chemical transformations for PET depolymerization, offering a promising approach for sustainable polymer waste management (Scheme 3, SI-6 and SI-7).
 | ||
| Scheme 3 Aminolysis and glycolysis using micronized PET powders. | ||
As mentioned, we performed the bead mill treatment using toluene. The resulting slurry was separated into two fractions: one that passed through a 500 µm mesh and one that was retained. The micronized PET for subsequent reactions was obtained from the slurry by solvent removal. This procedure is crucial for reproducibility. Since the micronized PET in the slurry was largely insoluble, it is possible to recover it by filtration rather than evaporation. We successfully demonstrated that micronized PET could be recovered using a 4 µm pore size filter paper, and the separated toluene could be reused (SI-8).
Bead mill-driven structural changes and contribution to methanolysis
This section discusses how bead milling affects PET structure and the mechanism of methanolysis, focusing on the role of PET crystallinity. To avoid the influence of impurities in the standard reagent PET, we primarily used supplier-provided amorphous and crystalline PET pellets, with used beverage PET flakes as a reference.Fig. 4 shows the 4-hour methanolysis results for these samples. The methanolysis rate for the amorphous PET pellet (PET B) was faster than for the crystalline PET pellet (PET C). Time-on-stream profiles (Fig. 5) showed that the crystalline PET pellet was gradually converted to DMT, reaching approximately 90% yield after 16 hours, while the amorphous and used beverage PET powders degraded rapidly. To understand this difference, we analyzed the samples using Differential Scanning Calorimetry (DSC) (Fig. 6a–c). Only the amorphous PET pellet showed both an exothermic peak (due to crystallization during heating) and a melting peak, with an estimated initial “crystallinity”65 of 7.9%. The other two samples showed only a melting peak above 200 °C, with crystallinities of 30.2% for the used beverage PET flakes and 40.1% for the crystalline PET pellet (Table 6).
 | ||
| Fig. 5 Time course of the methanolysis (2); effect of PET samples red circles, the amorphous PET pellet; green circles, the crystalline PET pellet; blue circles, the used beverage PET flake. Yields were determined by GC analyses. | ||
 | ||
| Fig. 6 Results of DSC analyses for three PET samples and changes caused by bead mill treatment. | ||
| Entry | PET sample | Bead-milling | T M [°C] | Melting enthalpy [mJ mg−1] | Crystallinity [%]a |
|---|---|---|---|---|---|
| a Melting enthalpy/140.1 × 100, see ref. 58. b Melting enthalpy–Post-crystallization enthalpy. | |||||
| 1 | Amorphous, pellet | No | 260.5 | 11.0b | 7.9 |
| 2 | Amorphous, powder | Done | 257.7 | 55.8 | 39.8 |
| 3 | Crystalline, pellet | No | 244.5 | 56.2 | 40.1 |
| 4 | Crystalline, powder | Done | 255.6 | 51.5 | 36.8 |
| 5 | Used beverage, flake | No | 252.1 | 42.4 | 30.2 |
| 6 | Used beverage, powder | Done | 253.0 | 50.9 | 36.3 |
Interestingly, bead milling influenced these thermal behaviors (Fig. 6d–f). For the amorphous PET pellet, the exothermic crystallization peak disappeared, suggesting that crystallization occurred during milling. For the used beverage PET flakes, a slight increase in crystallinity suggested crystallization of amorphous regions. This transformation from amorphous to crystalline phases can be explained by localized heat generation from collision energy, which induces crystallization in the polymer's secondary structure. Given that crystalline PET is less reactive, this increase in crystallinity is unlikely to be the primary cause of the observed higher reactivity of the amorphous PET. Instead, it should be considered a secondary result of the increased reactivity.
Bead milling, which involves simultaneous heating and solvent cooling, likely causes amorphous regions to undergo repeated expansion and contraction. This could strain the polymer's secondary structure, making chains more susceptible to cleavage. After bead milling, the crystallinity of all samples converged to approximately 35%, regardless of their initial crystallinity. It is known that extended-chain crystals tend to form under pressurized conditions, rather than the more stable folded-chain crystals.16,66,67 In particular, under mechanochemical conditions, the degree of crystallinity is reported to converge to around 30% owing to an equilibrium between the high and low crystallinity PET.66 Although the influence of physical factors such as localized heat and pressure in such mechanochemical processes remains a matter of debate, the present results are consistent with the reported tendency. On the other hand, our findings indicate that changes in crystallinity and molecular weight reduction occur simultaneously, suggesting they are interdependent phenomena.
Table 7 shows the physical property changes after 3 hours of bead milling. Notably, the primary particle size of the PET pellets (entries 2, 4, 6) was relatively large compared to the reagent PET pellet, which consists of aggregates with a reinforcer. A comparison of Mn and Mw for the amorphous and crystalline PET pellets after 3 hours of milling revealed that the decrease in Mn was more pronounced for amorphous PET. This indicates that while the degradation rate of high molecular weight polymer was similar, there were differences in the degradation behavior of the intermediate molecular weight region. Scanning electron microscopy (SEM) images of the bead-milled samples confirmed surface melting and elongation, indicating local exposure to temperatures above the melting point. These findings demonstrate that dynamic changes from physical stimuli like bead milling contribute to chemical changes in the polymer (SI-3-3). While we hypothesize that radicals play a key role in polymer cleavage, direct evidence is still under investigation.
| Entry | PET sample | Bead-millinga | Particle [µm]b | Molecular weight distributionc | Methanolysis resultd | |||
|---|---|---|---|---|---|---|---|---|
| M n | M w | M w/Mn | M p | |||||
| a Performed under standard conditions. b Determined by DLS analysis. c Determined by GPC analysis. d Results of methanolysis reaction under the standard conditions. e M(powder)/M(mother sample) × 100. | ||||||||
| 1 | Amorphous, pellet | No | — | 17400 |
62100 |
3.6 | 65800 |
<1 |
| 2 | Amorphous, powder | Done | 36 | 5600 (32e) | 31600 (51e) |
5.6 | 17300 |
84 |
| 3 | Crystalline, pellet | No | — | 15200 |
79300 |
5.2 | 77700 |
<1 |
| 4 | Crystalline, powder | Done | 51 | 6765 (45e) | 37300 (47e) |
6.6 | 12200 |
64 |
| 5 | Used beverage, flake | No | — | 26400 |
75900 |
2.9 | 74100 |
2 |
| 6 | Used beverage, powder | Done | 34 | 12100 (46e) |
43500 (57e) |
3.6 | 41400 |
86 |
We next focused on tracking the dynamic changes in the polymer during bead milling and methanolysis. The GPC profiles for amorphous and crystalline PET pellets, pulverized in toluene with zirconia beads for different times, are shown in Fig. 7 (results for used beverage PET flakes are in the SI).
 | ||
| Fig. 7 Changes over time in GPC analysis during bead mill treatment of (a) the amorphous PET pellet and (b) the crystalline PET pellet. | ||
For the amorphous PET pellet (Fig. 7a), the GPC profile after 1 hour of milling showed a shift in the peak of the maximum molecular weight toward a lower molecular weight. After 3 hours, the profile had two distinct peaks: a larger molecular weight peak (Peak H) as a shoulder and a new peak (Peak L) with a peak-top (Mp) of 17300 (Table 7, entry 2). This mid-range peak continued to shift to the oligomer region (3000 to 1000) after 18 to 48 hours of milling. In contrast, Peak H, which was a shoulder, remained in the same position but became a minor component after 6 hours.
A similar trend was observed for crystalline PET (Fig. 7b). The original peak at Mp = 77700 split into two distinct peaks after 3 hours of milling. A new mid-range peak, Peak L, appeared with an Mp of 12200. Like the amorphous PET, the high molecular weight component (Peak H) was prominent initially but became a minor component after 6 hours. These observations are consistent with the molecular weight distribution analysis. Although there were slight differences in the behavior of Peak H, its near-disappearance after 6 hours in both cases suggests that higher molecular weight fractions degrade relatively quickly. The intermediate molecular weight component (Peak L, Mp ≈ 10000–20000) showed an even more dramatic shift toward lower molecular weights after 3 hours. In contrast, components below Mw ≈ 10000 degraded much more slowly from 18 to 48 hours. For crystalline PET, a peak-top around 2000 remained detectable even after 24 hours of milling. This suggests that during bead milling, high-to mid-molecular weight PET chains degrade rapidly, but chains with molecular weights below approximately 10000 degrade much more slowly. This observation aligns well with the discussion on RED values based on HSP.
To investigate the mechanism of chemical bond cleavage during bead milling, we performed ESI-MS analysis (SI-9). When the amorphous PET pellet was bead-milled for 18 hours under standard conditions, we detected relatively small molecules with m/z = 2700 (Fig. 7a). These oligomers were small enough for ESI-MS analysis. Measurements in negative ion mode showed fragmentation patterns for a PET oligomer with a repeating structure, likely having a carboxylate group at one end and an ethylene glycol group at the other. This suggests that bead milling induces cleavage of the ester C–O bonds within the PET structure. The carboxylic acid termini of these oligomers could act as catalytic inhibitors in base-catalyzed depolymerization, which might explain the decrease in DMT yield observed with extended bead milling times.
We also performed bead milling in methanol for 24 hours and analyzed the resulting material. In addition to the previously observed oligomer, we detected another oligomer with a smaller mass number (m/z = 30). This structure is likely a phthalic acid monomethyl ester derivative with an ethylene glycol terminus. These findings suggest that during bead milling in methanol, PET undergoes C–O bond cleavage and simultaneously reacts with methanol. While this molecular weight reduction is often attributed to the solvent's wetting effect, these results also imply that solvents like methanol may directly participate in the bond cleavage reaction.
To gain insight into the dynamic changes of the insoluble polymer during methanolysis, we continued GPC analysis. When a crystalline PET pellet was bead-milled for 3 hours, a new peak (Peak L, Mw ≈ 12200) appeared alongside the original high molecular weight peak (Peak H, Mw ≈ 51300) (Fig. 7b). We then subjected this micronized sample to methanolysis and collected the insoluble PET components by filtration after 1, 3, and 10 hours for GPC analysis (Fig. 8).
 | ||
| Fig. 8 Changes over time in GPC analysis during methanolysis reaction of PET powder obtained from the crystalline PET pellet. a Reaction time of methanolysis. | ||
Within 1 hour of methanolysis, Peak L disappeared, and a new peak (Peak S, Mw ≈ 2900) appeared. Simultaneously, Peak H shifted and formed a shoulder (Peak M, Mw ≈ 28000). After 3 hours, the intensity of Peak M decreased significantly, and Peak S was also markedly reduced. By 10 hours, Peak H persisted with reduced intensity, while Peaks M and S had disappeared. These results were obtained by tracking insoluble PET components. Their disappearance means they were either solubilized or depolymerized over time. This indicates that small molecular weight PET was rapidly consumed during methanolysis, while high molecular weight PET persisted and gradually depolymerized into lower molecular weight species. This dynamic behavior during methanolysis is a stark contrast to what we observed during bead milling.
Although PET is mostly insoluble in methanol, analysis of the bead-milled PET that was soluble in methanol under heating revealed a component with a molecular weight of approximately 1000 in the solution phase. The absence of peaks in the high molecular weight region suggests that Peak S (Mw ≈ 2900) represents the lower molecular weight threshold of the insoluble PET component. Further analysis of the soluble fraction after 3 hours of methanolysis showed a single sharp peak in the monomer region (Fig. 9), indicating that PET oligomers that transition into the homogeneous phase are rapidly solvolyzed into monomers.
 | ||
| Fig. 9 GPC analysis of insoluble and soluble PET fragments during methanolysis reaction. | ||
Based on our GPC investigation, the PET depolymerization process proceeds via the following mechanism:
(1) Rapid degradation of high molecular weight PET during bead milling, with the degradation rate decreasing as molecular weight becomes smaller.
(2) Relatively fast depolymerization of mid-to low-molecular weight components during methanolysis. These components converge to PET oligomers with Mp = 2,900, which are then rapidly converted into soluble PET species.
(3) The soluble PET is rapidly solvolyzed to produce the monomer DMT.
Recirculating continuous bead mill treatment and one-pot milling-methanolysis process
Having detailed the molecular weight reduction of PET by bead milling and its subsequent depolymerization, we now discuss our efforts to leverage the advantages of the bead mill method.A key feature of bead milling is its capacity for continuous pulverization. By continuously feeding a slurry into a milling space, the process can be scaled up without a large vessel. To demonstrate this, we used a horizontal continuous bead mill (details in SI). The device operates in circulation mode, consisting of a slurry receiver, a continuous bead mill vessel with a 0.09 mm slit, and a pump. We periodically monitored the slurry's pulverization state and incrementally added 5.0 g of used beverage PET flakes at 4-hour intervals once the target micronization level was reached. This setup produced 17.4 g of PET powder (Fig. 10). The continuously micronized PET was then depolymerized at 90 °C, yielding DMT with a high yield of 91% (SI-11-1).
 | ||
| Fig. 10 Continuous bead mill treatment. | ||
While previous studies successfully evaluated bead milling and depolymerization separately, integrating them into a one-pot system is a novel and significant research direction. This approach simplifies the overall process and effectively uses the heat generated during milling to drive the reaction. The simultaneous execution of solvolysis and pulverization represents a highly efficient conversion method.
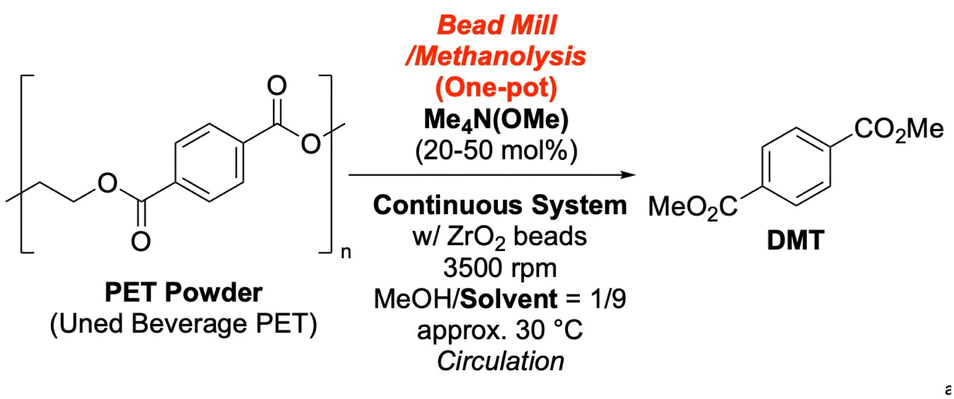
To implement a one-pot process, we first conducted a batch reaction using 1.0 g of used beverage PET flakes in a MeOH/pyridine (1:9) solvent system with 10 mol% TMAM. This yielded DMT in a satisfactory 72% yield after 6 hours (Scheme 4 and SI-10). We then investigated a continuous bead mill setup. We introduced 1.0 g of PET and 20 mol% TMAM into a 1 L MeOH/pyridine (1:9) solvent system and circulated the slurry. Analysis of the reservoir solution showed a DMT yield of 30% after 1 hour, with no significant improvement after 24 hours. The larger solvent volume in the continuous setup, compared to the batch process, likely contributed to the lower reaction rate due to dilution.
 | ||
| Scheme 4 One-pot bead mill/methanolysis in batch system. | ||
Increasing the catalyst loading to 50 mol% did not improve the yield and even caused a reduction, suggesting that factors beyond dilution were at play. Interestingly, pyridine has a low Hansen solubility parameter-based RED value (0.37),58–61 suggesting it might dissolve PET during milling. This dissolution would be detrimental to molecular weight reduction, which is critical for depolymerization efficiency. Substituting pyridine with toluene as a co-solvent dramatically improved efficiency, achieving quantitative conversion of PET to DMT within 2 hours. This result is comparable to our earlier finding that chloroform as a bead milling solvent produced highly reactive PET powder, despite minimal molecular weight reduction. Although the poor results with pyridine may seem contradictory to the chloroform findings, the role of co-solvents is complex. For example, dichloromethane (DCM, RED = 0.06)58,59,61 was ineffective as a co-solvent in batch one-pot reactions, despite its known ability to dissolve PET. This highlights the significant role of molecular weight reduction in the one-pot process (Table 8 and SI-11-2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent system | Yield of DMT [%]a | |||||
| 1 | 3 | 5 | 8 | 20 | 24 | ||
| a Yield at the corresponding recirculation time [h]. Determined by 1H NMR analysis. | |||||||
| 1 | MeOH/pyridine = 1:9 (TMAM: 20 mol%) |
30 | 31 | 32 | 34 | 40 | 32 |
| 2 | MeOH/pyridine = 1:9 (TMAM: 50 mol%) |
10 | 10 | 10 | 10 | 11 | 11 |
| 3 | MeOH/toluene = 1:9 (TMAM: 50 mol%) |
56 | >99 | >99 | >99 | >99 | >99 |
Importantly, the methanol/toluene solvent system also gave a sufficient DMT yield of 70% in batch one-pot reactions, demonstrating the approach's versatility. This mechanochemical process, which combines bead milling and chemical reaction, is a unique and promising method for PET depolymerization. The lack of previous reports on the catalytic conversion of untreated PET to monomers at room temperature within such a short timeframe underscores the novelty of this methodology.36,68
Continuous-flow synthesis of tamibarotene from DMT
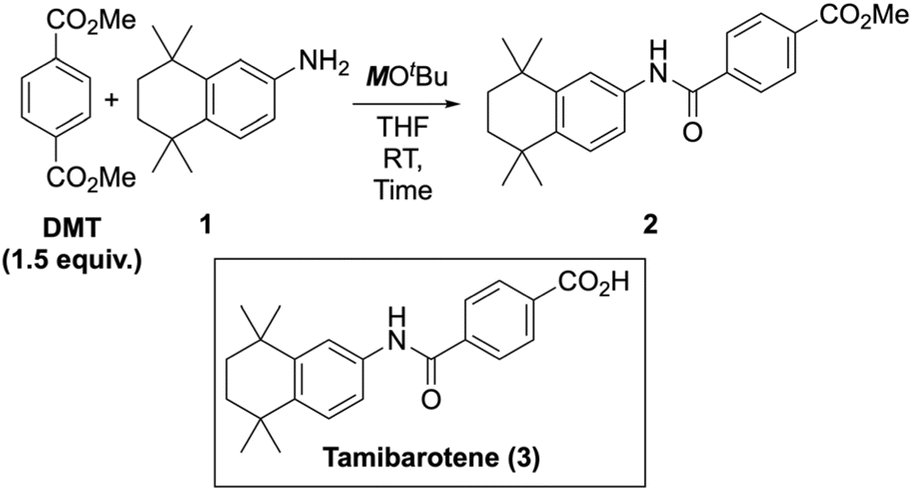
Current plastic recycling strategies face challenges from high costs44 and a mismatch between the volume of waste plastics and the market demand for upgraded materials.11 To address this, it is crucial to create diverse supply chains for high-value chemicals. Examples include converting PET to 1,4-benzenedimethanol (BDM)69–72 and dimethyl 1,4-cyclohexanedicarboxylate (DMCD)73via hydrogenolysis, and to terephthalamides via aminolysis.12,74,75 A particularly attractive solution for creating a cost-effective process is to convert recycled materials into high-value pharmaceuticals.In this study, we aimed to demonstrate the synthesis of the active pharmaceutical ingredient (API) tamibarotene (3) from DMT. Tamibarotene, marketed as Amnolake® in Japan, is used to treat ATRA-resistant acute promyelocytic leukemia (APL).76–78 While tamibarotene synthesis from monomethyl terephthalate (MMT) via an enzymatic approach was recently reported,79 a more straightforward method involves the amidation of an aniline derivative (1) with terephthalic acid monochloride.80,81 We explored an ester-amide exchange reaction using potassium tert-butoxide (KOtBu) as a base promoter. Focusing on a continuous-flow system, we used a THF solution of the base.
Under batch conditions, using 2.0 equiv. of KOtBu at 90 °C for 6 hours yielded the desired amide (2) in 80% isolated yield (Table 9, entry 2). By reducing the temperature to room temperature and shortening the reaction time, we effectively suppressed side reactions and achieved a nearly quantitative yield of 2 within 1 hour (entry 7). Lowering the amount of KOtBu to 1.5 or 1.0 equiv. reduced the yield but allowed the reaction time to be shortened to 3 hours. Substituting KOtBu with NaOtBu or LiOtBu led to progressively lower yields (SI-12).
|
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Time [h] | Conv. [%]a | Yield [%]a |
| a Determined by 1H-NMR analysis. b The reaction was performed at 90 °C. c Isolated yield. d The reaction was performed at 60 °C. e 1.2 Equiv. of DMT was employed. | ||||
| 1b | KOtBu (2.0) | 24 | >99 | 65 |
| 2b | KOtBu (2.0) | 6 | >99 | 86 (80c) |
| 3d | KOtBu (2.0) | 6 | >99 | 97 |
| 4 | KOtBu (2.0) | 6 | >99 | 99 |
| 5 | KOtBu (2.0) | 3 | >99 | 99 |
| 6e | KOtBu (2.0) | 3 | 89 | 83 |
| 7 | KOtBu (2.0) | 1 | 97 | 95 (94c) |
| 8 | KOtBu (1.5) | 6 | 93 | 90 |
| 9 | KOtBu (1.0) | 6 | 58 | 58 |
| 10 | NaOtBu (2.0) | 1 | 59 | 56 |
| 11 | LiOtBu (2.0) | 1 | 5 | 3 |
| 12 | KOMe (2.0) | 1 | 1 | Trace |
We then applied these conditions to a continuous-flow system (Fig. 10). In the initial trial, Reservoir 1 contained a 0.1 M THF solution of amine 1 and KOtBu, and Reservoir 2 contained a 0.15 M THF solution of DMT. The solutions were pumped at 0.13 mL min−1, merged, and directed into a 10 m tube reactor (1.0 mm internal diameter). With a total flow rate of 0.26 mL min−1, the residence time was 30 minutes. However, the pump clogged after 10 minutes due to the precipitation of potassium salts at the connector. Since a potassium-based base is necessary for the reaction, preventing inorganic precipitate formation was a challenge.
To solve this, we investigated mixed solvent systems with highly polar solvents such as DMSO, DMF, NMP, and tBuOH. A 4:1 DMSO/THF mixture for Reservoir 1 provided the most promising results by improving solution transparency and reducing the likelihood of precipitation (SI-12-3). While this flow mode initially gave a 20% yield of amide 2, the yield was not sustainable and dropped to 5% after 3 hours. We also observed that aniline 1 gradually degraded in the basic solution of Reservoir 1. To address this, we modified the system so that Reservoir 1 contained a mixture of DMT and amine 1, while Reservoir 2 delivered only KOtBu. We also used a sonicator (commonly used lab equipment) to prevent occasional precipitation within the reactor tube. This modified system worked well, allowing the reaction to proceed smoothly and achieve a nearly quantitative yield of the amide product (Fig. 11). Under these conditions, the productivity was calculated to be 0.6 mmol h−1, and the space–time yield was determined to be 3.5 g h−1 dL−1.
 | ||
| Fig. 11 Continuous-flow synthesis of tamibarotene precursor. | ||
To identify the structure of the precipitate. 1H NMR analysis of the white precipitate, collected under argon, showed peaks corresponding to the amide structure, with the aromatic protons of the aniline moiety shifted to a higher field. ESI-MS showed a dominant peak corresponding to the mass of 2-H (m/z = 364.19), suggesting the precipitate was a potassium amide formed by the deprotonation of the product (amide 2). In the continuous-flow study, the outlet solution was quenched with acetic acid to prevent further reaction. We also observed in a batch reaction that quenching with water induced hydrolysis of the ester, quantitatively converting the product into tamibarotene. These findings indicate that our continuous-flow system is a promising methodology for the efficient synthesis of tamibarotene from recycled PET-derived intermediates.
Conclusions
In conclusion, we have identified several catalytic depolymerization systems for PET that operate at temperatures below 90 °C. In all cases, wet pulverization via bead mill treatment proved to be a critical enabling technology for low-temperature depolymerization. The molecular weight reduction achieved during the bead milling step, which serves as the first stage of our process, plays a vital role in facilitating the low-temperature methanolysis in the second stage. We discovered that this molecular weight reduction is largely explained by the affinity between the solvent and PET. We also found that the transition from an amorphous to a crystalline phase, induced by localized heating, significantly influences PET's reactivity. This provided a crucial explanation for the observed differences in reactivity based on the initial PET properties. By tracking the degradation behavior during both the bead milling and methanolysis steps, we elucidated a mechanism in which lower molecular weight components are depolymerized into monomers more rapidly. Ultimately, we achieved PET monomerization without external heating by combining continuous bead mill treatment with methanolysis. Furthermore, we demonstrated a proof-of-concept for upcycling by continuously converting the resulting DMT into pharmaceuticals.A striking finding from this study is the crucial role of solvents, which was greater than we initially anticipated. These effects were observed only under wet pulverization conditions using the bead mill method, highlighting its unique capabilities. While this study focused on PET depolymerization as a key milestone, we are now exploring the application of this methodology to the chemical recycling of various other polymers.
Author contributions
T. K. conducted all experimental works. H. I. and S. K. designed and directed the project. All authors were involved in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in supporting information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc06930k.Acknowledgements
Bead milling was performed in our laboratory using the AIMEX EasyNano RMB II for batch system, and AIMEX Start Lab RMH for continuous system. The authors convey their appreciation to Mr Yasuo Igarashi and Mr Akihiro Igarashi of AIMEX Co., Ltd for technical support with machinery, and discussions. The authors also acknowledge Mitsui Chemicals Inc. for helpful discussions and GPC measurements. The authors also acknowledge Dr Tei Maki of JEOL Ltd for microscopic analysis. This work was supported by Grant-in-Aid for Scientific Research from the New Energy and Industrial Technology Development Organization (NEDO, JPNP 19004) project and JSPS KAKENHI Challenging Research, Exploratory (Grant 23K17362), Japan.Notes and references
- American Chemical Society, Bakelite: The World's First Synthetic Plastic, http://www.acs.org/content/acs/en/education/whatischemistry/landmarks/bakelite.html, accessed Jan 28, 2025.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 25 Search PubMed.
- Our World in Data, Global plastics production, https://ourworldindata.org/grapher/global-plastics-production, accessed Jan 28, 2025.
- M. Dokl, A. Copot, D. Krajnc, Y. Van Fan, A. Vujanović, K. B. Aviso, R. R. Tan, Z. Kravanja and L. Čuček, Sustain. Prod. Consum., 2024, 51, 498 CrossRef CAS.
- P. S. Roy, G. Garnier, F. Allais and K. Saito, ChemSusChem, 2021, 14, 4007 CrossRef CAS PubMed.
- J. Araujo-Morera, R. Verdejo, M. A. López-Manchado and M. Hernández Santana, Waste Manag., 2021, 126, 309 CrossRef PubMed.
- M. Antoniadou, T. Varzakas and I. Tzoutzas, Circ. Econ. Sustain., 2021, 1, 563 CrossRef PubMed.
- G. Gaustad, M. Krystofik, M. Bustamante and K. Badami, Resour. Conserv. Recycl., 2018, 135, 24 CrossRef.
- M. R. Karimi Estahbanati, X. Y. Kong, A. Eslami and H. S. Soo, ChemSusChem, 2021, 14, 4152 CrossRef CAS PubMed.
- L. T. Korley, T. H. I. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803 CrossRef CAS PubMed.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659 CrossRef CAS.
- E. Luna, I. Olazabal, M. Roosen, A. Müller, C. Jehanno, M. Ximenis, S. de Meester and H. Sardon, Chem. Eng. J., 2024, 482, 148861 CrossRef CAS.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041 CrossRef CAS PubMed.
- C. Bai, R. J. Spontak, C. C. Koch, C. K. Saw and C. M. Balik, Polymer, 2000, 41, 7147 CrossRef CAS.
- S. Molina-Boisseau and N. Le Bolay, Chem. Eng. J., 2000, 79, 31 CrossRef CAS.
- C. Basire, US pat., 20130150514A1, 2013 Search PubMed.
- E. G. Ashton, W. Kindlein, R. Demori, L. H. A. Cândido and R. Mauler, J. Clean. Prod., 2016, 116, 268 CrossRef CAS.
- I. Sánchez-Calderón, V. Bernardo, D. Cuadra-Rodríguez, J. Martín-de-León and M. Á. Rodríguez-Pérez, Polymer, 2022, 261, 125397 CrossRef.
- R. GmbH, Application database, https://www.retsch.com/applications/application-database/, accessed Jan 28, 2025.
- V. Chipakwe, P. Semsari, T. Karlkvist, J. Rosenkranz and S. C. A. Chelgani, J. Mater. Res. Technol., 2020, 9, 8148 CrossRef CAS.
- K. Kushimoto, A. Kondo, T. Kozawa, M. Naito and J. Kano, Adv. Powder Technol., 2024, 35, 104689 CrossRef.
- A. Kwade and J. Schwedes, KONA Powder Part. J., 1997, 15, 91 CrossRef CAS.
- F. P. Bowden, A. D. Yoffe and P. W. Levy, Phys. Today, 1959, 12, 58 CrossRef.
- P.-A. Thiessen, K. Meyer, G. Heinicke, Grundlagen Der Tribochemie, De Gruyter, Berlin, 1967 Search PubMed.
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC.
- T. Ogi, R. Zulhijah, T. Iwaki and K. Okuyama, KONA Powder Part. J., 2017, 34, 3 CrossRef CAS.
- R. GmbH, Bead Mills, https://www.retsch.com/products/bead-mills/, accessed Jan 28, 2025.
- H. Zhao, Y. Ye, Y. Zhang, L. Yang, W. Du, S. Wnag and Z. Hou, Chem. Commun., 2024, 60, 13832 RSC.
- J. Zhou, T.-G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300768 CrossRef CAS PubMed.
- I. R. Speight, K. J. Ardila-Fierro, J. G. Hernández, F. Emmerling, A. A. Michalchuk, F. García, E. Colacino and J. Mack, Nat. Rev. Methods Primers, 2025, 5, 29 CrossRef CAS.
- V. Štrukil, ChemSusChem, 2021, 14, 330 CrossRef PubMed.
- A. W. Tricker, A. A. Osibo, Y. Chang, J. X. Kang, A. Ganesan, E. Anglou, F. Boukouvala, S. Nair, C. W. Jones and C. Sievers, ACS Sustainable Chem. Eng., 2022, 10, 11338 CrossRef CAS.
- S. Gai, Y. Li, Y. Wang, Z. Guo, B. Liu, L. Huang, J. Beiyuan, D. Liu, R. Cha and W. Yuan, Chem. Eng. J., 2024, 500, 157131 CrossRef.
- D. Jain, F. Cramer, P. Shamaraienko, H.-J. Drexler, B. Voit and T. Beweries, RSC Sustain., 2025, 3, 3513 RSC.
- S.-C. Kosloski-oh, Z. A. Wood, Y. Manjarrez, M. W. Fieser and Z. A. Wood, Mater. Horiz., 2021, 8, 1084 RSC.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347 CrossRef CAS.
- H. Luo, H. Tyrrell, J. Bai, R. I. Muazu and X. Long, Green Chem., 2024, 26, 11444 RSC.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8 CrossRef CAS.
- M. Wang, Y. Li, L. Zheng, T. Hu, M. Yan and C. Wu, Polym. Chem., 2024, 15, 585 RSC.
- M. H. Ghasemi, N. Neekzad, F. B. Ajdari, E. Kowsari and S. Ramakrishna, Environ. Sci. Pollut. Res., 2021, 28, 43074 CrossRef CAS PubMed.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185 CrossRef CAS.
- T. Kawase, H. Ishitani and S. Kobayashi, Chem. Lett., 2023, 52, 745 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and V. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127 CrossRef CAS.
- R. Abe, N. Komine, K. Nomura and M. Hirano, Chem. Commun., 2022, 58, 8141 RSC.
- J. L. Hedrick, P. Kilickiran, G. W. Nyce, and R. M. Waymouth, US pat., 20040127720A1, 2004 Search PubMed.
- P. Mckeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721 RSC.
- A. El Mahdi, R. Medimagh, and T. Fourdin, WO pat., 2023194442A2, 2023 Search PubMed.
- S. Tanaka, J. Sato and Y. Nakajima, Green Chem., 2021, 23, 9412 RSC.
- S. Tanaka, M. Koga, T. Kuragano, A. Ogawa, H. Ogiwara, K. Sato and Y. Nakajima, ACS Mater. Au, 2024, 4, 335 CrossRef CAS PubMed.
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511 RSC.
- H. Essaddam, WO pat., 2017007965A1, 2017 Search PubMed.
- B. R. Debruin, A. A. Naujokas, and W. J. Gamble, US pat., 5432203A, 1995 Search PubMed.
- L. Li, Y. Zhang, X. Cui, Z. Said, S. Sharma, M. Liu, T. Gao, Z. Zhou, X. Wang and C. Li, J. Manuf. Process., 2023, 102, 921 CrossRef.
- S. Grätz and L. Borchardt, RSC Adv., 2016, 6, 64799 RSC.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, edn. 2nd edn, CRC Press, Boca Raton, 2007 Search PubMed.
- B. A. Pulido, O. S. Habboub, S. L. Aristizabal, G. Szekely and S. P. Nunes, ACS Appl. Polym. Mater., 2019, 1, 2379 CrossRef CAS.
- The Official Site for Hansen Solubility Parameters and HSPiP software. https://www.hansen-solubility.com/accessed Jan 28, 2025.
- S. S. Karim, S. Farrukh, T. Matsuura, M. Ahsan, A. Hussain, S. Shakir, L. F. Chuah, M. Hasan and A. Bokhari, Chemosphere, 2022, 307, 136050 CrossRef CAS PubMed.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606 CrossRef CAS.
- Y. Zhang, F. Tian, Z. Wu, X. Li, X. Liu and Y. He, Mater. Today Commun., 2022, 32, 104045 CrossRef CAS.
- J. Huang, D. Yan, Q. Zhu, X. Cheng, J. Tang, X. Lu and J. Xin, Polym. Degrad. Stab., 2023, 208, 110245 CrossRef CAS.
- S. Giraldo-Narcizo, N. Guenani, A. M. Sánchez-Pérez and A. Guerrero, ChemBioChem, 2023, 24, e202200503 CrossRef CAS PubMed.
- A. Zaker and K. Auclair, ChemSusChem, 2024, e202401506 Search PubMed.
- A. Misra and R. S. Stein, J. Polym. Sci. Polym. Phys. Ed, 1979, 17, 235 CrossRef CAS.
- H. W. Lee, K. Yoo, L. Borchardt and J. G. Kim, Green Chem., 2024, 26, 2087 RSC.
- E. M. Krall, T. W. Klein, R. J. Andersen, D. S. Reader, B. C. Dauphinais, S. P. McIlrath, A. A. Fischer, M. J. Carney and N. F. Robertson, Chem. Commun., 2014, 50, 4884 RSC.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem. – Eur. J., 2015, 21, 10851 CrossRef CAS PubMed.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- Y. Hu, S. Zhang, J. Xu, Y. Liu, A. Yu, J. Qian and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202312564 CrossRef CAS PubMed.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202117205 CrossRef CAS PubMed.
- B. M. Stadler and J. G. de Vries, Philos. Trans. R. Soc., A, 2021, 379, 20200341 CrossRef CAS PubMed.
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić and J. M. Modak, Adv. Mater., 2022, 34, 2100843 CrossRef CAS PubMed.
- I. Miwako and H. Kagechika, Drugs Today, 2007, 43, 563 CrossRef CAS PubMed.
- H. F. M. Nakagomi, M. Yamagata, H. Katsuki, K. Kawahara, K. Kitaoka, Y. Miki and K. Shudo, Biol. Pharm. Bull., 2012, 35, 1206 CrossRef PubMed.
- A. Takeshita, K. Shinagawa, M. Adachi, T. Ono, T. Kiguchi and T. Naoe, Expert Opin. Orphan Drugs, 2014, 2, 961 CrossRef CAS.
- E. Goulding, L. C. Ward, F. E. Allan, D. Dittman, J. E. Salcedo-Sora and A. J. Carnell, Angew. Chem., Int. Ed., 2025, 64, e202414162 CrossRef CAS PubMed.
- Y. Hamada, I. Yamada, M. Uenaka, T. Sakata, US pat., 005214202A, 1993 Search PubMed.
- X. Bao, X. Qiao, C. Bao, Y. Liu, X. Zhao, Y. Lu and G. Chen, Org. Process Res. Dev., 2017, 21, 748 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |