
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the application potential of AlphaFold3-like approaches in virtual screening
Chao
Shen
 *abc,
Xujun
Zhang
bd,
Shukai
Gu
bd,
Odin
Zhang
bd,
Qinghan
Wang
bd,
Gang
Du
bd,
Yihao
Zhao
bd,
Linlong
Jiang
bd,
Peichen
Pan
bd,
Yu
Kang
bd,
Qingwei
Zhao
ac,
Chang-Yu
Hsieh
abd and
Tingjun
Hou
*abd
*abc,
Xujun
Zhang
bd,
Shukai
Gu
bd,
Odin
Zhang
bd,
Qinghan
Wang
bd,
Gang
Du
bd,
Yihao
Zhao
bd,
Linlong
Jiang
bd,
Peichen
Pan
bd,
Yu
Kang
bd,
Qingwei
Zhao
ac,
Chang-Yu
Hsieh
abd and
Tingjun
Hou
*abd
aDepartment of Clinical Pharmacy, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310003, China. E-mail: shenchao513@zju.edu.cn; tingjunhou@zju.edu.cn
bCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, China
cZhejiang Provincial Key Laboratory for Drug Evaluation and Clinical Research, Hangzhou, Zhejiang 310003, China
dZhejiang Provincial Key Laboratory for Intelligent Drug Discovery and Development, Jinhua, Zhejiang 321016, China
First published on 11th December 2025
Abstract
AlphaFold3 (AF3) has revolutionized the paradigm for protein–ligand complex structure prediction, yet its potential for structure-based virtual screening (VS) remains largely underexplored. Herein, we present a systematic assessment of AF3-like approaches for VS applications, using AF3, Protenix and Boltz-2 as representative models. Initial benchmarks on the well-established DEKOIS2.0 datasets demonstrate AF3's exceptional screening capability, driven solely by its intrinsic confidence metrics for compound ranking. While third-party scoring schemes do not improve efficacy, both AF3 and Protenix prove robust as pose generators. Further analysis reveals performance declines in three more challenging cases: progressive exclusion of chemically similar active ligands from test sets, evaluation on a novel GPCR dataset with limited structural representation in model training, and assessment on a subset of LIT-PCBA dataset whose inactive compounds were experimentally verified. Despite these limitations, these models consistently surpass conventional docking tools in accuracy in most cases. Pose analysis further indicates that most predictions adopt physically plausible conformations, albeit with minor structural artifacts. This study highlights the promise and current constraints of AF3-like methods in VS, offering practical insights for their deployment in modern drug discovery.
Introduction
The field of structural biology has undergone a transformative revolution with the advent of AlphaFold,1 an artificial intelligence (AI) solution developed by DeepMind for protein structure prediction. Compared to AlphaFold2 (AF2) primarily limited to predicting isolated protein structures, the latest iteration, AlphaFold3 (AF3),2 has marked a substantial leap forward by extending its applicability beyond proteins alone, covering a broader range of biomolecular systems, particularly in modeling protein–ligand complexes. This enhanced capability could potentially deepen our understanding of molecular recognition processes and even profoundly benefit the early drug discovery workflows, since deciphering protein–ligand interactions is continually a fundamental challenge in the context of structure-based drug design (SBDD).As a cornerstone technique in SBDD, structure-based virtual screening (SBVS) plays a pivotal role in modern drug discovery for identifying novel hit compounds.3 A typical SBVS campaign begins with a three-dimensional (3D) protein structure and a large compound library, employing computational molecular docking to prioritize compounds with optimal binding scores for subsequent experimental validation. In contrast to ligand-based approaches that rely on the principle of structural similarity implying bioactivity, SBVS can provide detailed insights into binding mechanisms from a 3D structure perspective, making it a more suitable strategy for scaffold hopping and identifying structurally diverse compounds. Nevertheless, while docking-based VS has demonstrated remarkable success over the past decades,4,5 inherent limitations in docking algorithms persist as critical bottlenecks for improving screening efficacy. These challenges primarily stem from inadequate coverage of pose sampling,6 inherent inaccuracies in scoring functions,7,8 and insufficient accounting for protein flexibility during simulations.9,10
The ability to directly predict protein–ligand complex structures positions emerging co-folding approaches like AF3 as compelling alternatives to conventional docking methods. By leveraging generative diffusion models, AF3 bypasses the exhaustive conformational sampling characteristic of traditional search algorithms, while its built-in confidence metrics provide reliable scoring for pose prioritization. Moreover, unlike conventional docking methods that typically treat protein structures as rigid entities, AF3's sequence-based prediction paradigm inherently mitigates the challenges posed by protein flexibility, offering a more robust solution for biomolecular interaction modeling. However, while these advancements have facilitated AF3's remarkably superior accuracy over specialized docking tools on some established datasets,2 the generalizability of this performance toward broader chemical space warrants further systematic investigation.
Since the first release of AF3 in 2024 and the subsequent emergence of open-source derivatives such as Chai-1,11 Boltz-1,12 Protenix,13 and Boltz-2,14 extensive efforts have been devoted to exploring their applicability across diverse biomolecular systems.15–23 These investigations have encompassed protein–ligand complexes,15–17 protein–protein interactions,18,19 protein–peptide systems,20,21 and even more challenging ternary systems such as those involving proteolysis-targeting chimeras (PROTACs)22 or molecular glues.23 While the assessment results consistently suggest that current models depend more on memorization from training data than on genuine physical understanding of molecular interactions, most analyses to date have focused primarily on direct pose reconstruction of crystalized entities. Crucially, the potential utility of these predicted complex structures for downstream applications such as binding affinity prediction and VS remains largely unexplored. A notable exception comes from a recent study demonstrating AF3's near-perfect enrichment performance in distinguishing covalent active ligands from property-matched decoys.24 Nevertheless, given that the datasets used there are predominantly composed of kinases due to the requirement for covalent binding, the observed results may be biased, as the crystal structures of kinases have been extensively resolved and are thus overrepresented in training data. Hence, further evaluations across more diverse scenarios are still necessary.
In this study, to figure out whether AF3-like approaches could be consistently applicable to VS, a comparable assessment was conducted using Protenix and AF3 as primary examples. The assessment was further supplemented with the recently developed Boltz-2, which includes a dedicated binding affinity prediction module, enabling direct affinity estimation alongside structural prediction. The analysis was first performed on DEKOIS2.0 dataset,25 a well-established benchmark that had been widely employed to evaluate the VS performance of both physics-based and AI-powered docking tools. In addition to simply estimating screening performance using the intrinsic confidence scores provided by AF3, Protenix and Boltz-2, we further explored whether integrating AF3-predicted complex structures with high-precision third-party rescoring methods could enhance enrichment. Notably, given that the proteins and ligands in DEKOIS might overlap with the training data of these models, which may introduce potential biases, we additionally curated the GPCRrecent dataset (Fig. S1, S2 and Table S1), comprising protein targets whose first-determined crystal structures were released exclusively after 2022. To mitigate bias introduced by artificially-generated decoys in above two datasets, we also retrieved a subset of widely-recognized LIT-PCBA dataset,26 where both the actives and inactives were experimentally verified. Using these two extra datasets, we further benchmarked the performance of such approaches with Protenix and Boltz-2 as representative methods. Our rigorous evaluation across multiple datasets demonstrates the considerable promise of AF3-like approaches in VS tasks, while also revealing opportunities for further optimization to enhance their utility in practical VS projects.
Results and discussion
Evaluation of AF3's built-in confidence scores on DEKOIS2.0 dataset
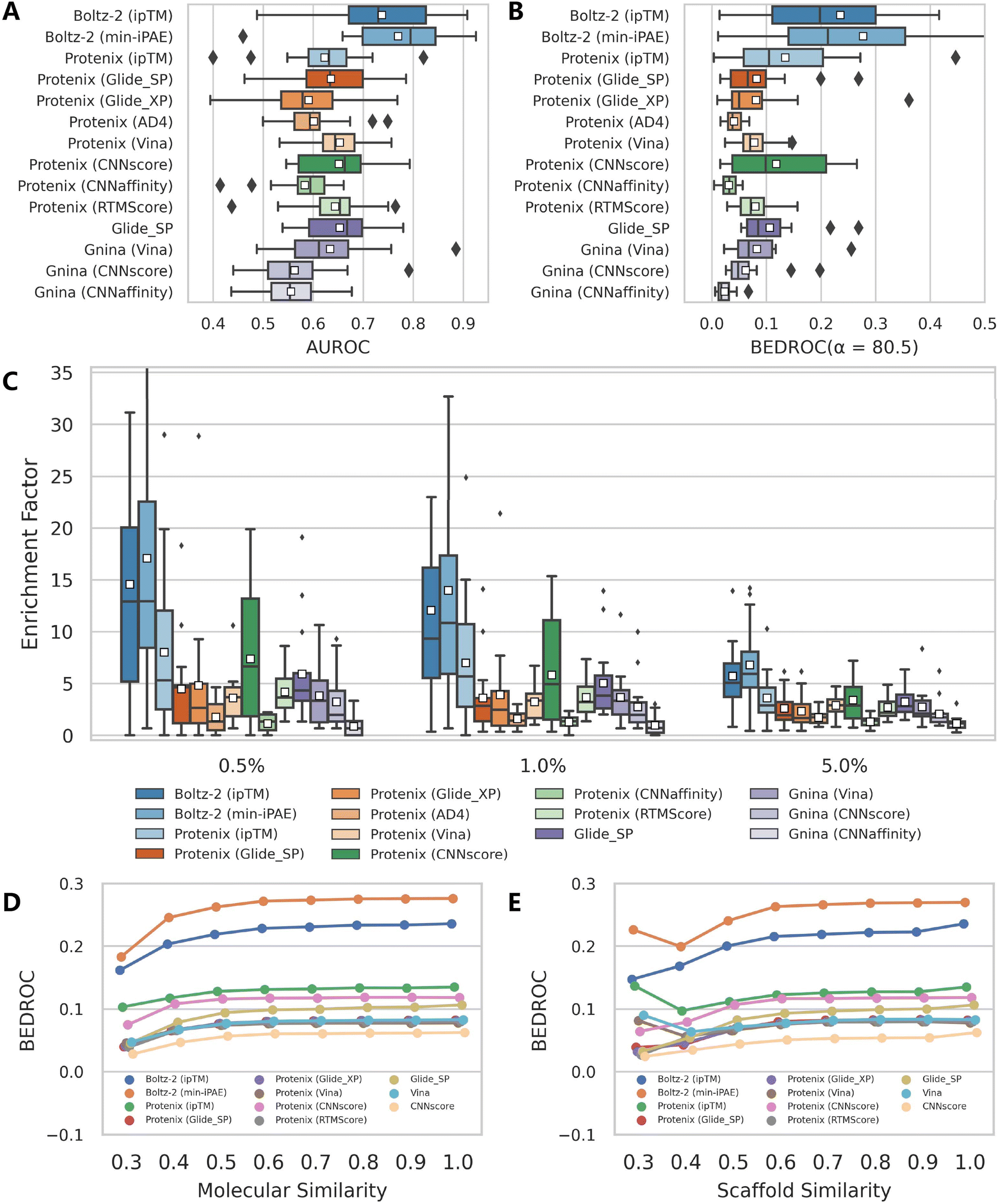
Using the well-established DEKOIS2.0 dataset, we first investigated the performance of Protenix, AF3 and Boltz-2 when directly coupled with their intrinsic confidence metrics. For comprehensive benchmarking, we compared these approaches with five popular traditional docking programs (Glide SP,27 AutoDock Vina,28 GOLD CHEMPLP,29 Surflex-Dock,30 and LeDock31) and five emerging AI-powered docking tools (Gnina,32 TankBind,33 KarmaDock,34 CarsiDock35 and SurfDock36). The comparative performance, indicated through AUROC, BEDROC, and enrichment factors (EFs), is detailed in Table 1 and Fig. 1.| Method | AUROC | BEDROC (α = 80.5) | EF0.5% | EF1% | EF5% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Med | Mean | Med | Mean | Med | Mean | Med | Mean | Med | |
| a As Boltz-2 directly fetched affinity data from public databases (e.g., PubChem, ChEMBL, and BindingDB), which also serve as the sources for DEKOIS 2.0, its reported performance metrics are likely significantly over-estimated due to this data overlap. Therefore, the results presented here should be treated as a reference only and may not reflect its true predictive performance. b The results were retrieved from ref. 38. c The results were retrieved from ref. 39. d The results were retrieved from ref. 34. e The results were retrieved from ref. 35. f The results were retrieved from ref. 36 and the indicators were calculated based on all 81 targets due to the unavailability of the label for each target. | ||||||||||
| AF3 (ranking score) | 0.893 | 0.934 | 0.628 | 0.655 | 20.57 | 22.14 | 19.23 | 21.46 | 12.42 | 13.50 |
| AF3 (pTM) | 0.832 | 0.874 | 0.417 | 0.396 | 13.68 | 13.29 | 12.59 | 11.92 | 9.34 | 9.50 |
| AF3 (ipTM) | 0.892 | 0.933 | 0.635 | 0.659 | 20.46 | 22.14 | 19.32 | 21.46 | 12.64 | 13.50 |
| AF3 (min-iPAE) | 0.913 | 0.952 | 0.755 | 0.828 | 22.98 | 26.57 | 23.15 | 26.23 | 14.50 | 16.00 |
| Boltz-2 (ranking score) | 0.831 | 0.857 | 0.385 | 0.347 | 12.89 | 13.29 | 11.77 | 9.54 | 7.89 | 7.50 |
| Boltz-2 (pTM) | 0.720 | 0.735 | 0.194 | 0.137 | 6.22 | 4.43 | 5.61 | 4.76 | 4.32 | 3.00 |
| Boltz-2 (ipTM) | 0.860 | 0.894 | 0.560 | 0.618 | 19.17 | 22.14 | 17.96 | 19.08 | 10.30 | 11.00 |
| Boltz-2 (min-iPAE) | 0.896 | 0.939 | 0.709 | 0.780 | 23.32 | 26.57 | 22.73 | 26.23 | 13.15 | 14.00 |
| Boltz-2 (Affinity)a | 0.854 | 0.918 | 0.705 | 0.780 | 25.17 | 31.00 | 23.54 | 28.59 | 11.66 | 12.50 |
| Boltz-2 (Probability)a | 0.911 | 0.964 | 0.820 | 0.925 | 26.68 | 31.00 | 25.84 | 28.62 | 14.32 | 16.00 |
| Protenix (ranking score) | 0.782 | 0.813 | 0.456 | 0.477 | 16.42 | 17.71 | 14.97 | 14.31 | 8.12 | 8.50 |
| Protenix (pTM) | 0.597 | 0.602 | 0.089 | 0.016 | 2.41 | 0.00 | 2.29 | 0.00 | 2.20 | 0.50 |
| Protenix (ipTM) | 0.795 | 0.837 | 0.507 | 0.553 | 18.44 | 22.14 | 16.81 | 16.69 | 8.84 | 9.50 |
| Glide_SPb | 0.745 | 0.752 | 0.374 | 0.313 | 14.20 | 13.23 | 12.01 | 9.53 | 6.18 | 5.95 |
| GOLD_CHEMPLPc | 0.647 | 0.631 | 0.172 | 0.148 | 5.78 | 4.43 | 5.38 | 4.75 | 3.36 | 3.00 |
| LeDockc | 0.656 | 0.680 | 0.187 | 0.142 | 6.78 | 4.43 | 5.88 | 4.77 | 3.65 | 3.50 |
| Surflex-Dockb | 0.671 | 0.673 | 0.219 | 0.180 | 8.30 | 4.43 | 7.27 | 4.77 | 3.97 | 3.50 |
| Gnina (Vina) | 0.653 | 0.659 | 0.164 | 0.105 | 6.17 | 4.43 | 5.16 | 2.38 | 3.12 | 2.50 |
| Gnina (CNNscore) | 0.715 | 0.734 | 0.354 | 0.342 | 13.73 | 13.29 | 11.92 | 9.54 | 6.17 | 6.00 |
| Gnina (CNNaffinity) | 0.710 | 0.739 | 0.257 | 0.231 | 9.36 | 8.86 | 8.21 | 7.15 | 4.87 | 4.50 |
| TankBindd | 0.602 | 0.606 | 0.109 | 0.053 | 2.90 | 0.00 | 2.94 | 0.00 | 2.42 | 1.51 |
| KarmaDock (Aligned)d | 0.743 | 0.786 | 0.458 | 0.453 | 16.55 | 17.45 | 15.16 | 15.16 | 7.33 | 7.01 |
| CarsiDocke | 0.797 | 0.838 | 0.561 | 0.638 | 20.23 | 22.14 | 18.65 | 23.85 | 9.29 | 9.00 |
| SurfDockf | 0.758 | 0.803 | 0.488 | 0.482 | 21.00 | 25.73 | 18.17 | 18.07 | 8.34 | 8.12 |
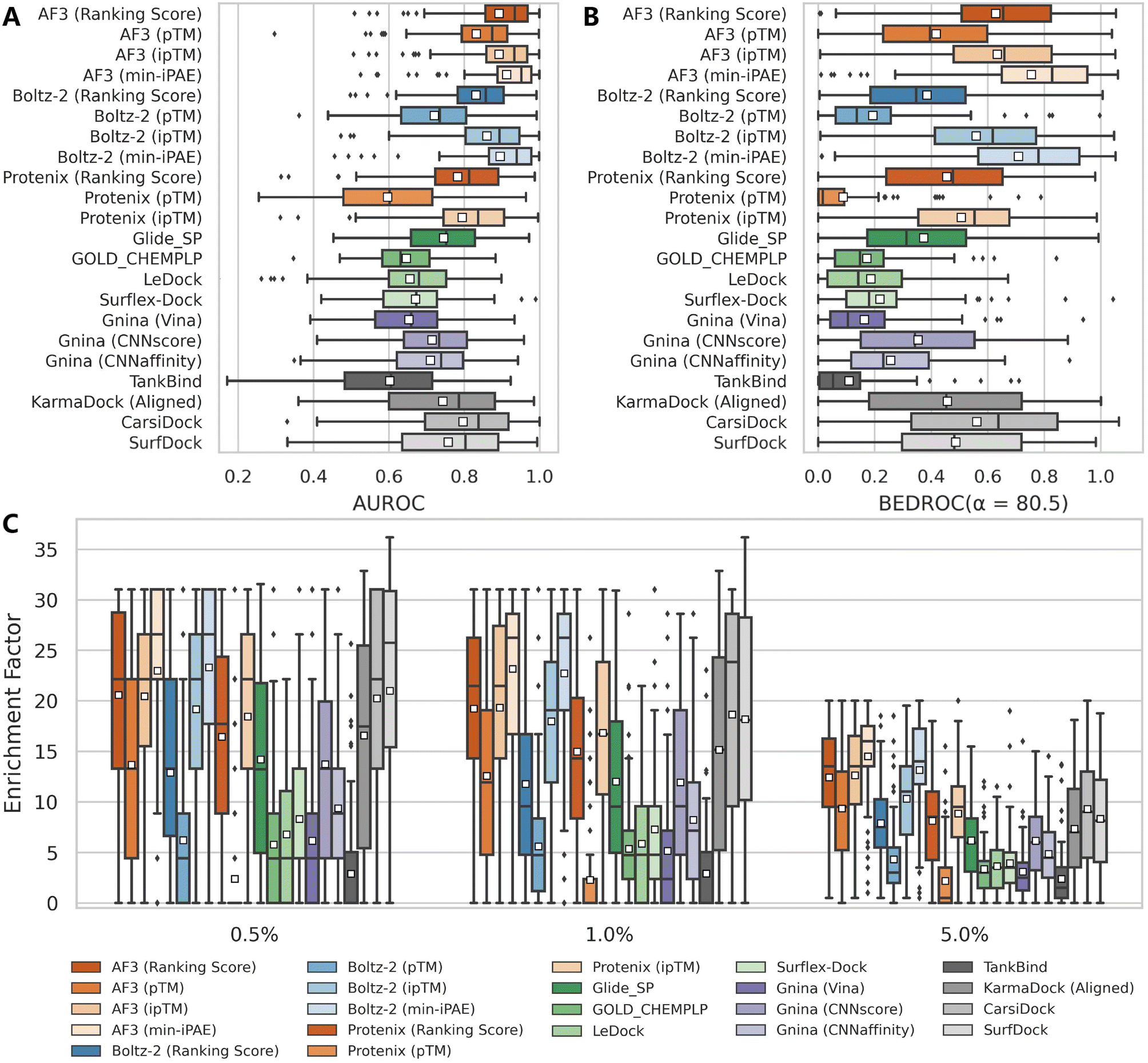 | ||
| Fig. 1 Performance comparison of multiple screening approaches on the DEKOIS2.0 benchmark set (N = 79), indicated by (A) AUROC, (B) BEDROC (a = 80.5) and (C) enrichment factors at thresholds of 0.5%, 1.0%, and 5.0%. White squares in box plots indicate mean values for each metric. The results for approaches except AF3, Protenix, Boltz-2 and Gnina were directly retrieved from previous studies,34–36,38,39 and the indicators for SurfDock were calculated based on all 81 targets due to the unavailability of the label for each target. | ||
Among the three confidence scores output from Protenix, ipTM exhibits the strongest discriminatory power, followed by Ranking score, while pTM performs significantly worse. This aligns with expectations, as ipTM specifically characterizes protein–ligand interfaces, whereas pTM primarily reflects global structural features. Ranking score, a composite metric integrating both pTM and ipTM, logically occupies an intermediate position. Notably, Protenix performs substantially inferior to Boltz-2 and AF3 across all confidence metrics and evaluation criteria in our assessment. While Protenix and Boltz-2 were developed as open-source implementations inspired by AF3, they likely differ in critical aspects such as training data scale, quality, and undisclosed methodological details from the original AF3 model. Furthermore, the development focus of Protenix may have prioritized overall structural accuracy over refining their built-in confidence metrics for VS. These findings suggest that further refinement may be still necessary for Protenix to match AF3's predictive capabilities for VS.
Despite so, Protenix (ipTM) still achieves competitive results, with a mean AUROC of 0.795, BEDROC of 0.507, EF0.5% of 18.44, EF1% of 16.81 and EF5% of 8.84, which not only markedly outperform those of the widely-employed traditional docking programs (the corresponding indicators of the best-performing Glide SP are 0.745, 0.374, 14.20, 12.01 and 6.18), but rival state-of-the-art AI-based approaches like CarsiDock (0.797, 0.561, 20.23, 18.65 and 9.29) and SurfDock (0.758, 0.488, 21.00, 18.17 and 8.34). The performance gap narrows considerably with AF3 (ipTM), which delivers metrics comparable to or even exceeding those of leading AI tools. Another interesting finding is that min-iPAE, a metric first proposed by Omidi et al.37 in AF-Multimer to capture interactions in intrinsically disordered protein regions, demonstrates even better performance than ipTM across all evaluated metrics for both AF3 (e.g., mean BEDROC: 0.755 vs. 0.635; EF1%: 23.15 vs. 19.32) and Boltz-2 (e.g., mean BEDROC: 0.709 vs. 0.560; EF1%: 22.73 vs. 17.96). This finding corroborates previous observations by Shamir et al.,24 highlighting its exceptional potential as an enrichment discriminator. As for the underlying mechanisms, we hypothesize both ipTM and min-iPAE may resemble that of knowledge-based scoring functions to distinguish actives from decoys. Unlike physics-based or empirical scoring functions that explicitly incorporate binding affinity data, these approaches leverage the structural reliability of predicted complexes for compound ranking, which may offer a promising avenue for further enhancing VS performance.
Regarding the marginal superiority of AF3 over Boltz-2 when evaluated using their native confidence metrics, one may attribute it to the difference in recommended number of predictions, i.e., the former outputs five models while the latter provides only one. To examine whether this discrepancy surely influenced the comparison, we conducted a simple experiment in which only the first generated sample from AF3 and Protenix was used as the final prediction, thereby simulating a single-sample scenario comparable to Boltz-2. As shown in Fig. S3, limiting the number of samples to one has only a minor impact on most evaluation metrics. In certain cases, models using only one prediction even slightly outperform those using multiple samples. These results suggest that it is methodologically acceptable to directly compare AF3 and Boltz-2 despite differences in their default sample numbers. Furthermore, they indicate that generating a single structural sample may be sufficient for large-scale VS, which has positive implications for computational efficiency in practical applications.
As expected, the affinity scores generated by the specialized binding affinity module in Boltz-2, from both its regression (Affinity) and classification (Probability) models, demonstrate exceptional enrichment performance (Table 1). However, it is important to note that Boltz-2 was trained on extensive affinity data sourced from public databases (e.g., PubChem,40 ChEMBL,41 and BindingDB42), which also serve as the primary sources for the active ligands in DEKOIS2.0 benchmark. As the Boltz-2 team has not released their specific training set, the actual degree of data overlap remains unquantifiable. Therefore, these results are presented for reference only and will not be incorporated into the following analyses in this study.
Further analysis of individual target performance (Fig. 2 and S4) reveals that while the four confidence scores embedded in AF3 exhibit nearly identical performance distributions in terms of AUROC, only Ranking score and ipTM show similar trends for other metrics. Interestingly, despite sharing similar prediction principles, Protenix, AF3 and Boltz-2 show divergent optimal targets under identical confidence metrics. This discrepancy becomes even more pronounced when comparing methods with fundamentally distinct screening protocols. Together, these observations emphasize the importance of target-specific evaluation when selecting or optimizing computational methods for a given protein system.
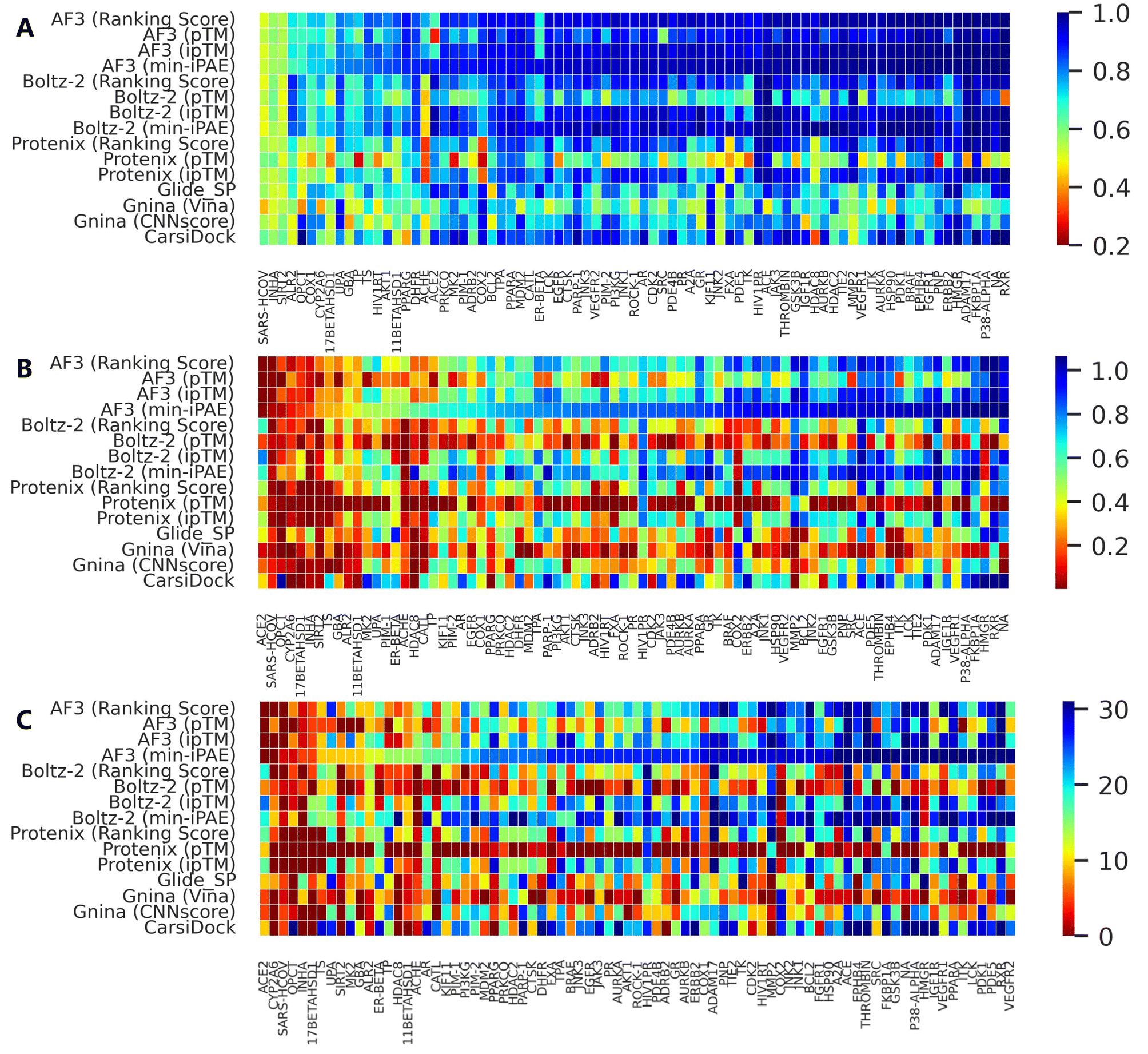 | ||
| Fig. 2 Performance distribution of several representative screening approaches across all targets in DEKOIS2.0 benchmark set (N = 79), evaluated by (A) AUROC, (B) BEDROC (a = 80.5) and (C) EF1%. Targets are sorted in ascending order based on the performance of AF3 (min-iPAE) for each individual metric. | ||
Impact of external scoring functions on screening performance
While AF3 combined with its intrinsic confidence scores has demonstrated encouraging VS performance on DEKOIS2.0 dataset, these scores may lack explicit physical interpretations of protein–ligand interactions. In practice, researchers might still prefer classical scoring schemes for compound prioritization. Therefore, leveraging the binding complexes predicted by Protenix and AF3, we further evaluated whether high-precision rescoring methods could potentially enhance enrichment. For comprehensive assessment, we examined scoring functions from two representative docking programs (Glide and Gnina) alongside several recently-developed deep learning approaches (PLANET,43 PIGNet2,44 RTMScore,38 and IGModel45) with varying mechanisms.As summarized in Table 2, Fig. 3 and S5, while Protenix alone could not match the performance of AF3 under identical conditions, the performance gap narrows when alternative scoring methods are applied, suggesting that Protenix indeed achieves comparable performance to AF3 in binding pose prediction. But among all rescoring protocols, only CNNscore and RTMScore could approach the high performance of ipTM and min-iPAE, whereas other methods lag significantly. This discrepancy likely stems from the shared design philosophy of CNNscore and RTMScore, both of which prioritize structural reliability for pose ranking, mirroring the mechanism of AF3's confidence metrics.
| Method | AUROC | BEDROC (α = 80.5) | EF0.5% | EF1% | EF5% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Med | Mean | Med | Mean | Med | Mean | Med | Mean | Med | |
| Protenix | ||||||||||
| ipTM | 0.795 | 0.837 | 0.507 | 0.553 | 18.44 | 22.14 | 16.81 | 16.69 | 8.84 | 9.50 |
| Glide_SP | 0.785 | 0.811 | 0.390 | 0.366 | 14.46 | 13.29 | 12.65 | 11.92 | 7.23 | 6.50 |
| Glide_XP | 0.792 | 0.817 | 0.394 | 0.371 | 14.74 | 13.29 | 12.68 | 11.92 | 7.38 | 7.00 |
| Gnina (AD4) | 0.624 | 0.607 | 0.151 | 0.108 | 5.38 | 4.43 | 4.77 | 2.38 | 3.08 | 2.50 |
| Gnina (Vina) | 0.682 | 0.678 | 0.186 | 0.167 | 6.84 | 4.43 | 5.80 | 4.77 | 3.58 | 3.50 |
| Gnina (Vinardo) | 0.751 | 0.767 | 0.259 | 0.225 | 8.80 | 4.43 | 8.15 | 7.15 | 5.08 | 4.50 |
| Gnina (CNNscore) | 0.846 | 0.890 | 0.515 | 0.597 | 18.11 | 22.14 | 16.69 | 19.08 | 9.34 | 10.00 |
| Gnina (CNNaffinity) | 0.748 | 0.774 | 0.310 | 0.303 | 11.44 | 8.86 | 10.26 | 9.54 | 5.72 | 5.50 |
| RTMScore | 0.852 | 0.909 | 0.640 | 0.759 | 21.02 | 26.57 | 20.37 | 23.85 | 11.62 | 13.00 |
| PLANET | 0.703 | 0.742 | 0.140 | 0.105 | 4.15 | 0.00 | 3.92 | 2.38 | 3.27 | 3.00 |
| PIGNet2 | 0.731 | 0.763 | 0.292 | 0.199 | 9.70 | 8.86 | 9.09 | 7.15 | 5.68 | 4.00 |
| IGModel (pkd) | 0.786 | 0.815 | 0.348 | 0.342 | 12.11 | 8.86 | 11.41 | 9.54 | 6.68 | 6.50 |
| IGModel (rmsd) | 0.787 | 0.838 | 0.287 | 0.279 | 8.30 | 4.43 | 8.54 | 7.15 | 6.11 | 6.00 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||||
| AF3 | ||||||||||
| ipTM | 0.892 | 0.933 | 0.635 | 0.659 | 20.46 | 22.14 | 19.32 | 21.46 | 12.64 | 13.50 |
| Min-iPAE | 0.913 | 0.952 | 0.755 | 0.828 | 22.98 | 26.57 | 23.15 | 26.23 | 14.50 | 16.00 |
| Glide_SP | 0.798 | 0.816 | 0.398 | 0.352 | 14.41 | 13.29 | 12.65 | 11.92 | 7.32 | 7.00 |
| Glide_XP | 0.796 | 0.827 | 0.391 | 0.394 | 13.73 | 17.71 | 12.53 | 11.92 | 7.43 | 7.00 |
| Gnina (AD4) | 0.636 | 0.647 | 0.157 | 0.103 | 5.10 | 4.43 | 4.92 | 2.38 | 3.14 | 2.50 |
| Gnina (Vina) | 0.704 | 0.705 | 0.194 | 0.160 | 7.06 | 4.43 | 5.92 | 4.77 | 3.92 | 3.50 |
| Gnina (Vinardo) | 0.757 | 0.766 | 0.270 | 0.239 | 9.19 | 8.86 | 8.45 | 7.15 | 5.21 | 4.50 |
| Gnina (CNNscore) | 0.855 | 0.896 | 0.509 | 0.549 | 18.11 | 22.14 | 16.51 | 19.08 | 9.25 | 10.00 |
| Gnina (CNNaffinity) | 0.757 | 0.779 | 0.323 | 0.269 | 11.94 | 8.86 | 10.50 | 9.54 | 5.98 | 5.50 |
| RTMScore | 0.865 | 0.906 | 0.674 | 0.787 | 22.42 | 26.57 | 21.34 | 26.23 | 12.34 | 13.00 |
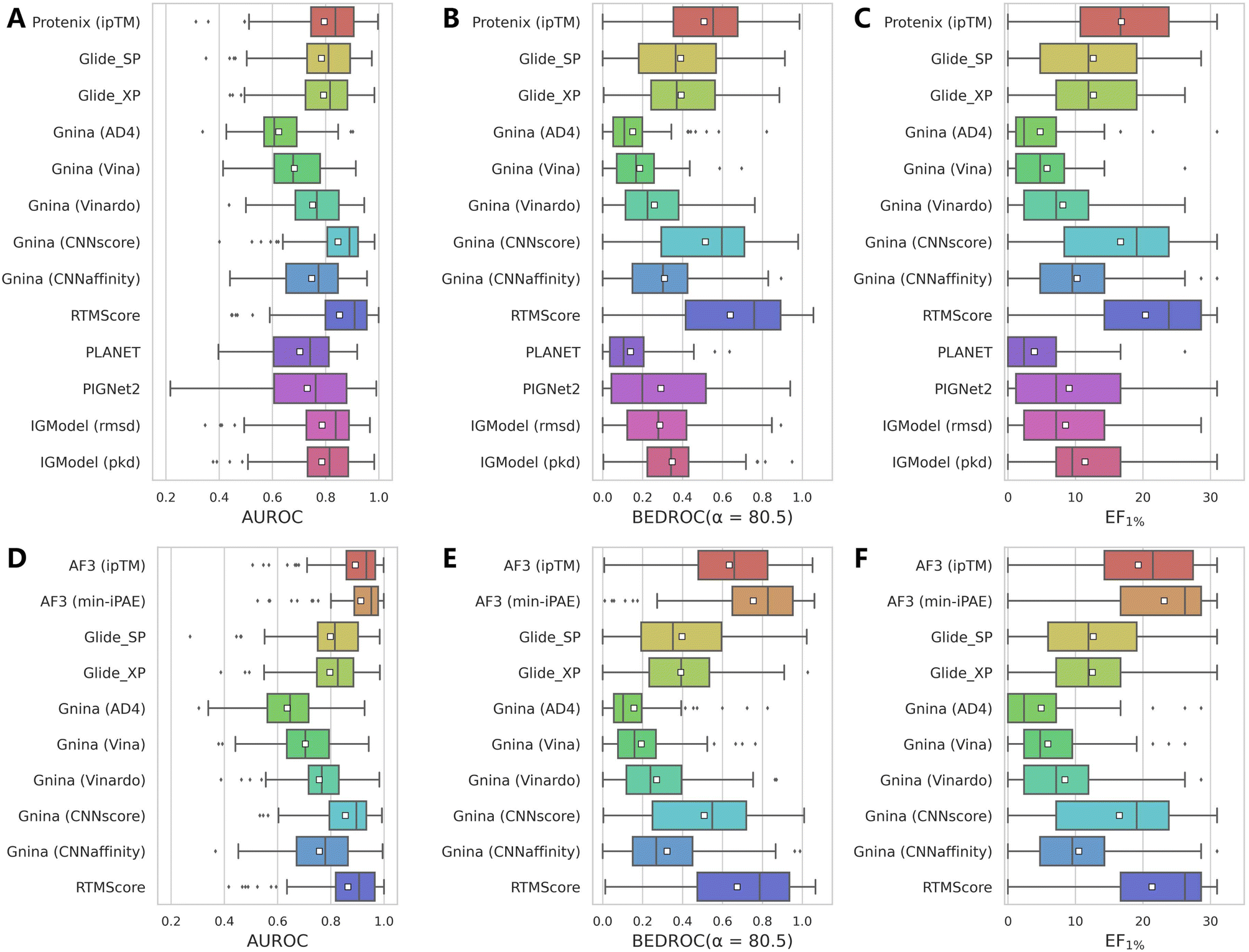 | ||
| Fig. 3 Performance comparison of multiple scoring approaches applied to structures predicted by (A–C) Protenix and (D–F) AF3 on the DEKOIS2.0 benchmark set (N = 79). Performance metrics include (A and D) AUROC, (B and E) BEDROC (a = 80.5) and (C and F) EF1%. White squares in box plots indicate mean values for each metric. | ||
To mitigate the influence of pose quantity on rescoring outcomes, we also compared scenarios involving all 5 candidate poses generated by the recommended settings of AF3/Protenix versus only the top-ranked pose selected by Ranking score. Fig. S6 and S7 depict the results for Protenix- and AF3-preidcted structures, respectively. Notably, CNNscore exhibits significant improvements, achieving metrics closely aligned with ipTM. Given that this model was trained on diverse cross-docked poses, its pronounced sensitivity to binding pose variations is unsurprising. In contrast, other methods show marginal gains or even performance degradation, suggesting that simply incorporating additional poses does not universally enhance outcomes.
We further investigated whether using AF3 or Protenix as alternative pose generators against traditional sampling algorithms (Glide/Gnina) could enhance VS performance, as detailed in Fig. 4A–C and S8. The results demonstrate consistent superiority of both AI-based generators over conventional methods in all tested scoring schemes, with AF3 showing a slight but negligible edge over Protenix. Notably, the performance enhancement is particularly significant for deep learning-based scoring functions (CNNscore, CNNaffinity, and RTMScore), with more modest improvements observed for classical methods (Glide SP and Vina). We also analyzed the pairwise correlation of each metric across the 79 targets using different pose generators with the same scoring scheme (Fig. 4D–I and S9). Intriguingly, the outcomes from AF3 and Protenix exhibit strong agreement, with Pearson's correlation coefficients (Rp) for BEDROC values ranging from 0.753 to 0.953. In contrast, comparisons between AF3/Protenix and classical search-based methods were significantly weaker (Rp values for AF3 and Protenix are 0.251–0.696 and 0.345–0.666, respectively). Additionally, AI-based scoring functions generally display higher correlations than classical ones, further supporting their reduced sensitivity to pose variations.
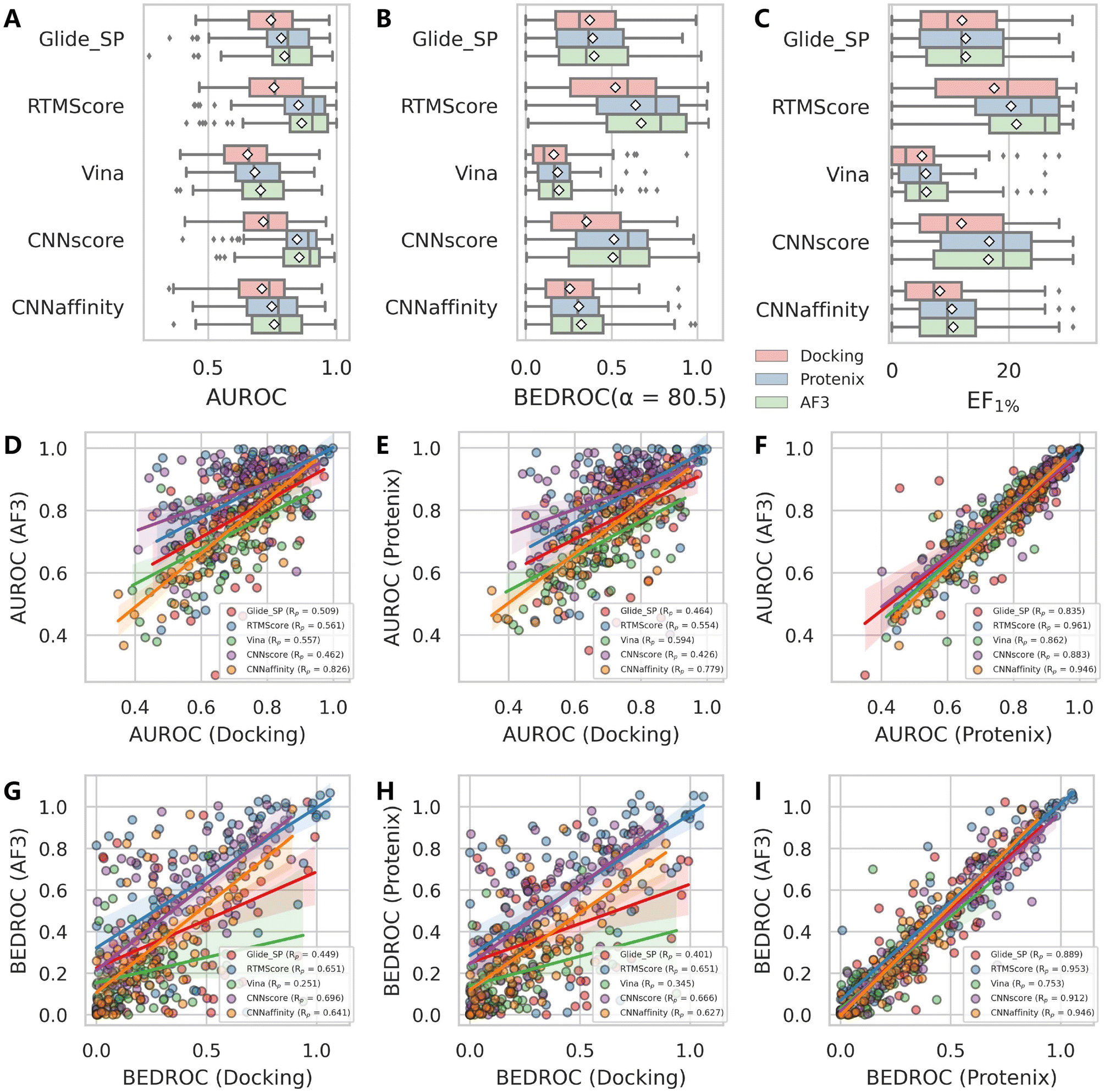 | ||
| Fig. 4 Evaluation of pose generation methods in screening performance. (A–C) Boxplots comparing the distributions of (A) AUROC, (B) BEDROC (a = 80.5) and (C) EF1% values across different pose generation methods. (D–I) Pairwise correlation analysis of (D–F) AUROC and (G–I) BEDROC values between different approaches: (D and G) Glide/Gnina vs. AF3, (E and H) Glide/Gnina vs. Protenix, and (F and I) AF3 vs. Protenix. The docking engine employed for RTMScore rescoring is Glide SP. White squares in box plots indicate mean values for each metric. | ||
Taken together, while none of the tested rescoring methods surpass AF3 (min-iPAE), RTMScore (mean AUROC: 0.865; BEDROC: 0.674; EF1%: 21.34) and CNNscore with 5 poses (mean AUROC: 0.877; BEDROC: 0.672; EF1%: 19.18) emerge as competitive alternatives. These findings also suggest the potential of AF3/Protenix as robust pose generators, particularly when paired with deep learning-based scoring approaches.
Impact of ligand similarity on screening performance
Although AF3 was not specifically optimized for binding affinity ranking, its training set presumably incorporated a substantial portion of protein–ligand complex structures available in the RCSB PDB.46 Therefore, it is highly likely that AF3 would assign high confidence scores for protein–ligand pairs that structurally resemble those encountered during training. While the protein structures in DEKOIS2.0 dataset may be overrepresented in AF3's training set, the vast chemical diversity of ligands makes comprehensive coverage improbable. We therefore systematically evaluated how ligand similarity affects screening performance on the DEKOIS2.0 dataset, irrespective of potential protein similarities. Our analysis began by retrieving all PDB ligands used in Protenix training and calculating their ECFP4 fingerprints,47 examining both entire molecules and more robust Murcko scaffolds.48 We then performed identical fingerprint calculations for each active compound in DEKOIS2.0 and thus determined their minimum Tanimoto similarity to any PDB ligand. For varying similarity thresholds, we re-calculated the performance metrics for each screening tool. Here we focused primarily on AUROC and BEDROC metrics because enrichment factors are particularly sensitive to the number of active compounds remaining after thresholding. As expected, when the retained active compounds exhibit strong enrichment, the overall performance in turn gains a significant improvement (Fig. S10).Fig. 5 reveals subtle yet discernible differences in performance trends between mean AUROC and BEDROC metrics. As ligand similarity decreases, AF3-like approaches combined with either ipTM or min-iPAE exhibit a noticeable progressive decline in AUROC scores, while other methods show only marginal variations until the similarity threshold drops below 0.4. Beyond this point, the exclusion of targets with insufficient qualifying active compounds introduces greater variability in the metrics. In contrast, the BEDROC metric presents a more consistent pattern, with all approaches following a similar downward trend. Notably, performance variations based on scaffold similarity are more pronounced than those observed for molecular similarity. These observations indicate that ligand similarity does influence the performance of AF3's intrinsic confidence scores to some degree. Importantly, despite these variations, the relative ranking among screening tools remains stable, with AF3 (min-iPAE) maintaining its superior performance even under low-similarity conditions. These trends were further accentuated in the median AUROC and BEDROC analyses (Fig. S11 and S12).
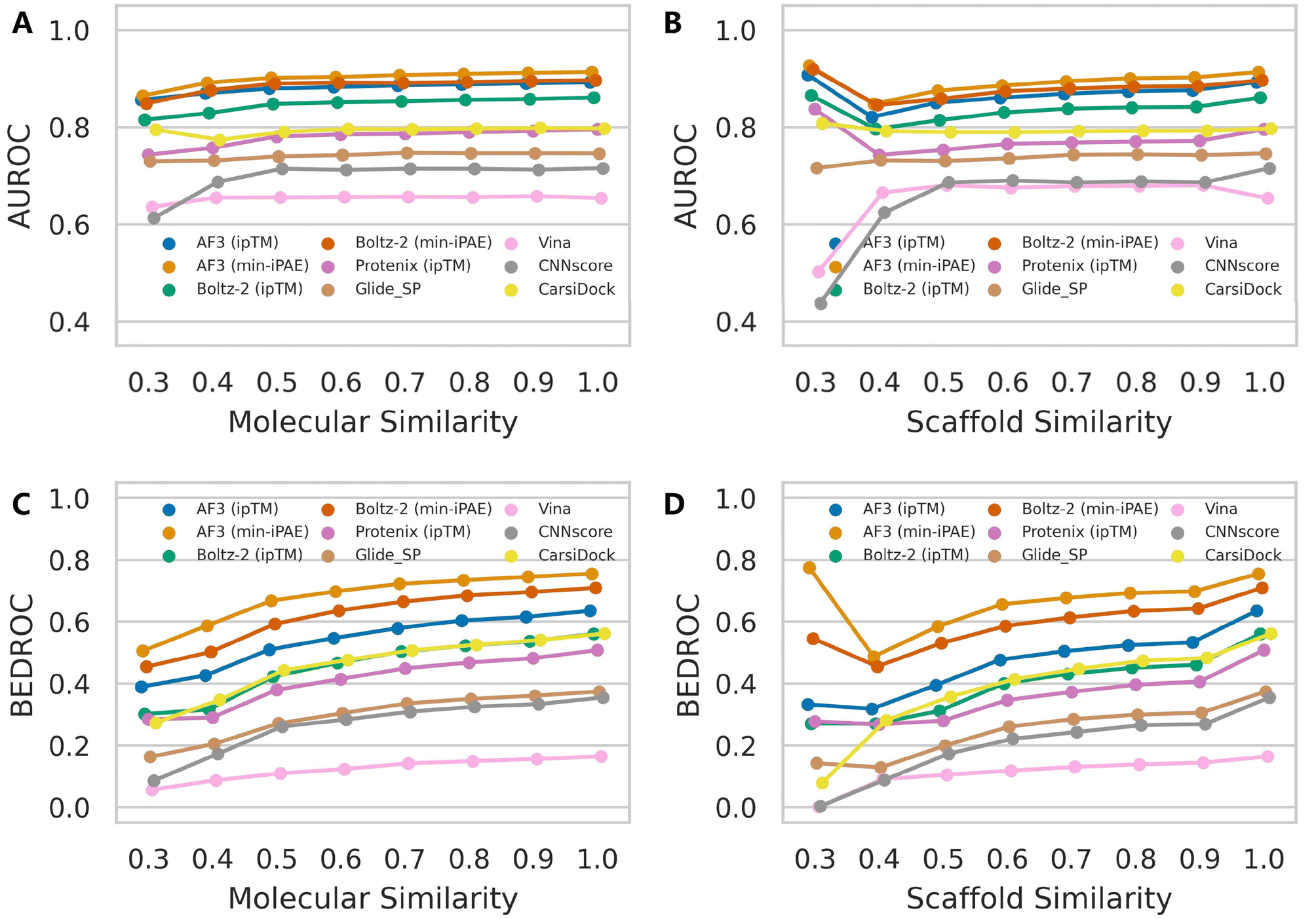 | ||
| Fig. 5 Impact of ligand similarity on the screening performance of multiple docking tools. Performance metrics include (A and B) AUROC and (C and D) BEDROC (a = 80.5). For each active compound in the DEKOIS2.0 dataset, ECFP4 fingerprints were computed either for (A and C) the entire molecule or (B and D) the Murcko scaffold. The minimum Tanimoto similarity to all ligands in the RCSB PDB was then determined. Data point represents the mean value across targets for each metric. Targets for which no active compounds met the specified similarity threshold were excluded from metric calculations. | ||
Similar patterns emerge when examining rescoring approaches (Fig. 6 and S13), with one key distinction: almost all methods exhibit a gradual decline in both mean AUROC and BEDROC as similarity decreases. Given that these rescoring schemes rely on structures predicted by either Protenix or AF3, this consistent trend suggests deteriorating pose generation accuracy for complexes featuring novel ligand scaffolds. Nevertheless, as clearly illustrated in Fig. 7, AF3- and Protenix-based pose generators consistently maintain superior performance compared to conventional search-based engines. This advantage is more substantial for deep learning-based CNNscore, whereas classical methods like Glide SP and Vina show progressively smaller performance gaps as similarity thresholds become more stringent.
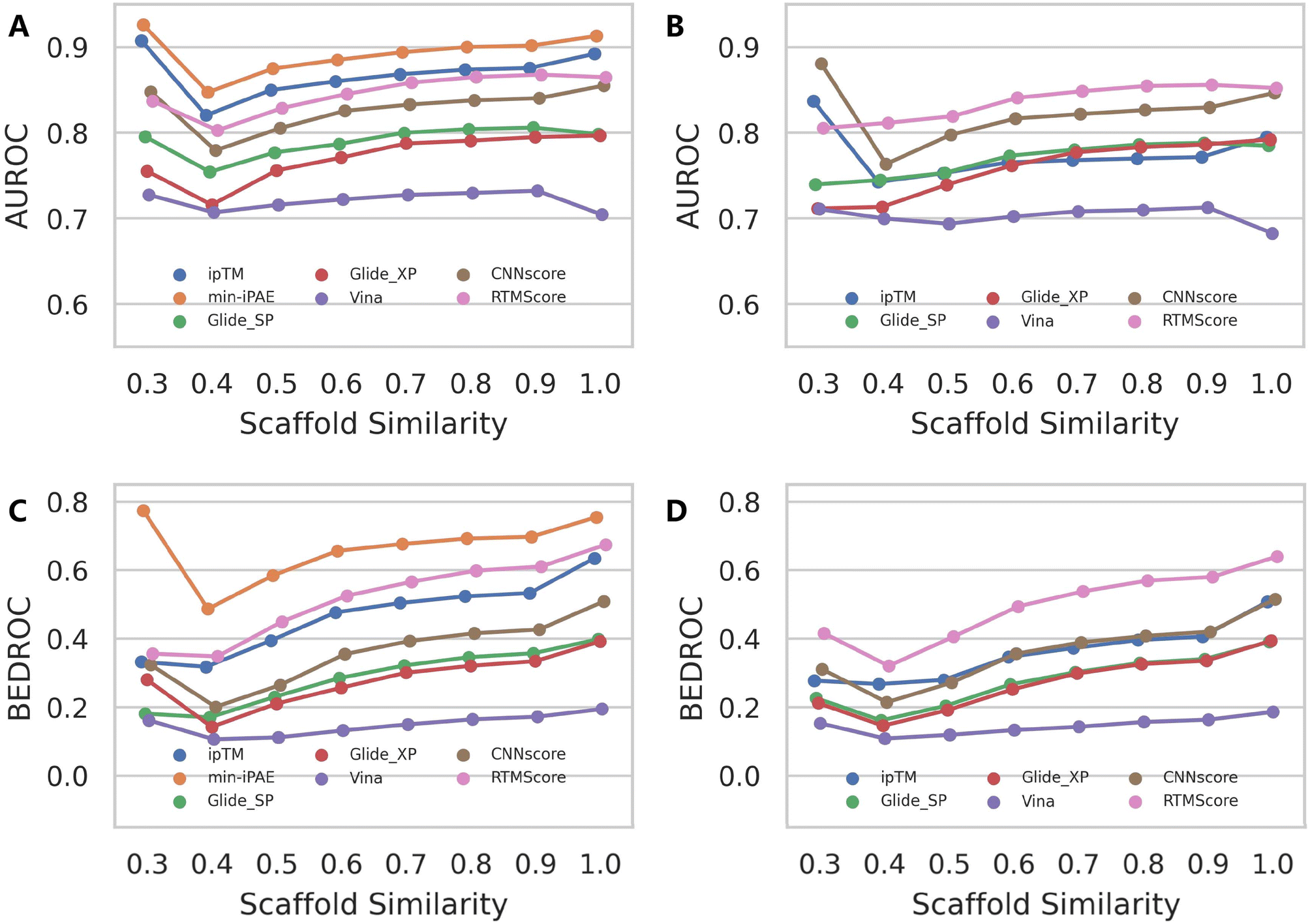 | ||
| Fig. 6 Impact of ligand similarity on the screening performance of multiple scoring schemes based on the structures predicted by (A and C) AF3 and (B and D) Protenix. Performance metrics include (A and B) AUROC and (C and D) BEDROC (a = 80.5). For each active compound in the DEKOIS2.0 dataset, ECFP4 fingerprints were computed for the Murcko scaffold. The minimum Tanimoto similarity to all ligands in the RCSB PDB was then determined. Data point represents the mean value across targets for each metric. Targets for which no active compounds met the specified similarity threshold were excluded from metric calculations. | ||
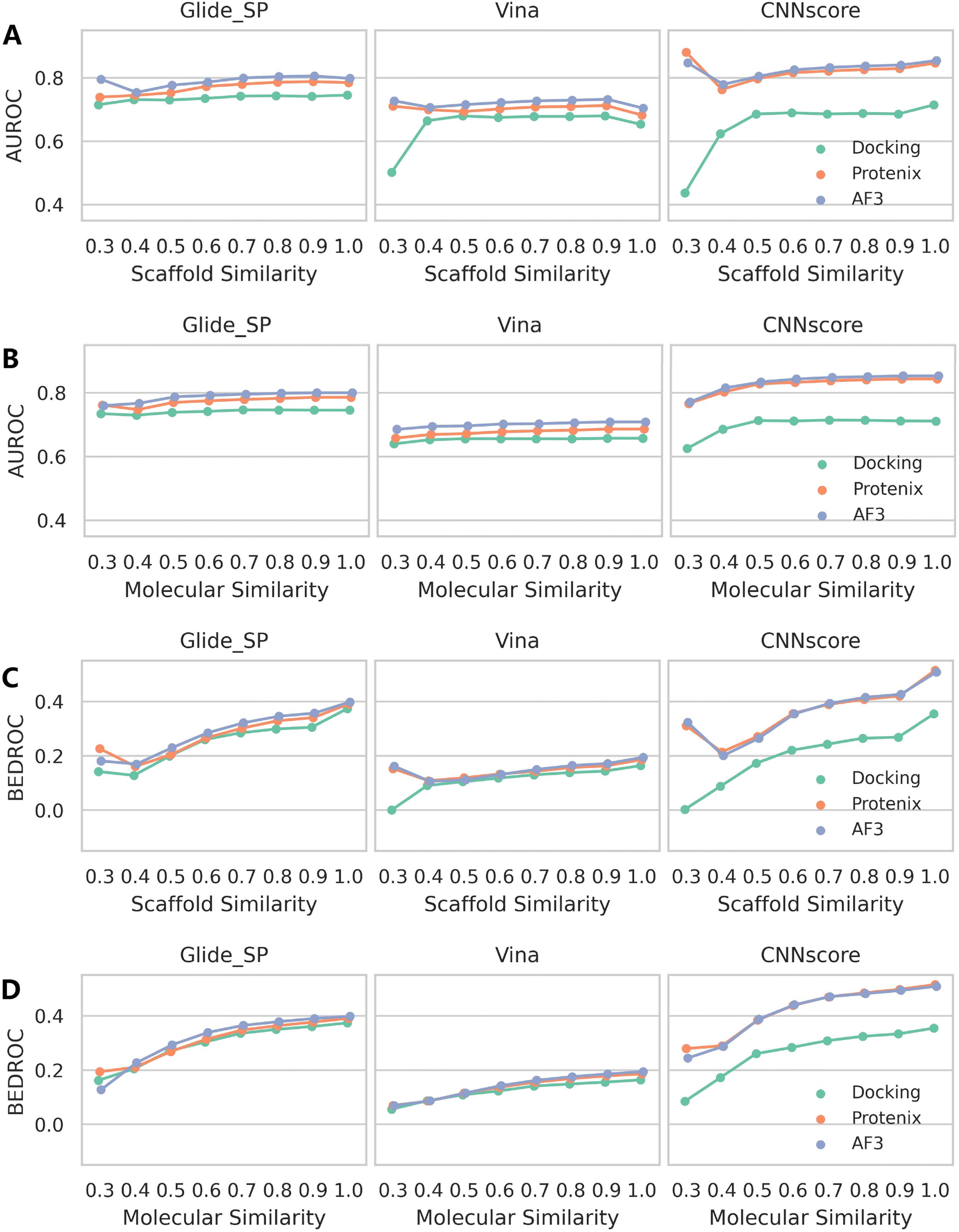 | ||
| Fig. 7 Impact of ligand similarity on screening performance across different rescoring schemes and pose generators. Performance metrics include (A and B) AUROC and (C and D) BEDROC (a = 80.5). For each active compound in the DEKOIS2.0 dataset, ECFP4 fingerprints were computed for (A and C) the entire molecule or (B and D) the Murcko scaffold. The minimum Tanimoto similarity to all ligands in the RCSB PDB was then determined. Data point represents the mean value across targets for each metric. Targets for which no active compounds met the specified similarity threshold were excluded from metric calculations. | ||
Performance on GPCRrecent dataset
To mitigate potential biases from overrepresented protein structures in model training, we further curated the GPCRrecent dataset, comprising only entries whose first crystal complex structures were released after 2022. We then evaluated the VS performance of AF3-like approaches, using Protenix and Boltz-2 as representative examples due to their significantly lower computational memory requirements compared to AF3. However, since Boltz-2 employed a temporal cutoff of 2023-6 in its training, we have to extract a corresponding subset containing only six targets for further analysis. Notably, the results yielded by its binding affinity prediction module were excluded from this particular analysis due to unavoidable data overlap between its training sources (e.g., BindingDB) and the composition of this benchmark. Consequently, while this temporally-constrained dataset may provide a complementary test for the methods primarily relying on crystal structures, it cannot adequately reflect the true generalization capability of those affinity prediction models like that in Boltz-2, which may have incorporated almost all available binding data into model training.The substantial differences in dataset composition and decoy generation mechanisms prevent direct generalization of DEKOIS2.0 benchmark results, as evidenced by Table 3 and Fig. 8. However, both Boltz-2 (min-iPAE) and Boltz-2 (ipTM) maintain strong performance, with mean BEDROC values of 0.236 and 0.277, and mean EF1% values of 12.03 and 13.96, respectively. These results remain consistent even when evaluated on a subset containing only six newer targets (0.230, 0.287, 12.62 and 14.30, respectively), though the limited number of targets precludes robust statistical conclusions. The metrics for Protenix (ipTM) lag significantly, but still demonstrates competitive early enrichment performance (mean BEDROC: 0.135, mean EF1%: 6.97) in comparison to the other approaches. Incorporating additional rescoring schemes does not yield significant improvements, with only CNNscore performing comparably (mean BEDROC: 0.118, mean EF1%: 5.80). Notably, as a pose generator, Protenix enhances performance only for CNNscore, likely due to its pronounced performance decline when using Gnina's built-in search engine, while results for Glide and Vina even deteriorate substantially. In contrast, conventional docking programs like Glide SP and Gnina (Vina) show reduced performance compared to their results on the DEKOIS2.0 dataset, yet their relative competence improves here. These observations imply that protein similarity indeed exerts a remarkable influence on those AI-driven tools, affecting both Protenix's complex structure prediction capability and the screening power of deep learning-based scoring functions.
| Method | AUROC | BEDROC (α = 80.5) | EF0.5% | EF1% | EF5% | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | Med | Mean | Med | Mean | Med | Mean | Med | Mean | Med | |
| a The results were obtained based on a subset of the whole dataset, which involves only the six targets whose first crystal structures released after 2023-6 (i.e., O15552, P0DMS8, P13945, P35348, P46098, Q96RJ0). | ||||||||||
| Boltz-2 (ipTM) | 0.738 | 0.730 | 0.236 | 0.198 | 14.56 | 12.91 | 12.03 | 9.30 | 5.70 | 5.03 |
| Boltz-2 (min-iPAE) | 0.770 | 0.795 | 0.277 | 0.213 | 17.06 | 12.91 | 13.96 | 10.83 | 6.78 | 5.93 |
| Boltz-2 (ipTM) subseta | 0.717 | 0.699 | 0.230 | 0.201 | 15.77 | 13.58 | 12.62 | 10.91 | 5.17 | 4.84 |
| Boltz-2 (min-iPAE) subseta | 0.764 | 0.740 | 0.287 | 0.245 | 19.03 | 17.55 | 14.30 | 12.50 | 6.83 | 5.87 |
| Protenix (ipTM) | 0.623 | 0.632 | 0.135 | 0.105 | 7.99 | 5.30 | 6.97 | 5.67 | 3.59 | 2.88 |
| Protenix (Glide_SP) | 0.636 | 0.634 | 0.082 | 0.066 | 4.47 | 4.64 | 3.58 | 2.83 | 2.59 | 1.93 |
| Protenix (Glide_XP) | 0.591 | 0.579 | 0.081 | 0.050 | 4.79 | 2.64 | 3.89 | 2.47 | 2.32 | 1.67 |
| Protenix (AD4) | 0.601 | 0.594 | 0.041 | 0.038 | 1.75 | 1.32 | 1.59 | 1.41 | 1.72 | 1.72 |
| Protenix (Vina) | 0.653 | 0.644 | 0.078 | 0.070 | 3.59 | 3.64 | 3.21 | 3.33 | 2.88 | 2.90 |
| Protenix (CNNscore) | 0.652 | 0.663 | 0.118 | 0.099 | 7.37 | 6.62 | 5.80 | 4.92 | 3.38 | 2.86 |
| Protenix (CNNaffinity) | 0.583 | 0.594 | 0.031 | 0.032 | 1.11 | 1.32 | 1.26 | 1.33 | 1.29 | 1.40 |
| Protenix (RTMScore) | 0.644 | 0.654 | 0.080 | 0.071 | 4.17 | 3.63 | 3.66 | 3.20 | 2.69 | 2.17 |
| Glide_SP | 0.653 | 0.668 | 0.106 | 0.085 | 5.90 | 4.30 | 5.02 | 3.83 | 3.22 | 2.80 |
| Gnina (Vina) | 0.635 | 0.612 | 0.083 | 0.067 | 3.80 | 3.31 | 3.65 | 3.33 | 2.73 | 2.10 |
| Gnina (CNNscore) | 0.564 | 0.554 | 0.062 | 0.048 | 3.18 | 1.99 | 2.74 | 1.97 | 2.06 | 1.73 |
| Gnina (CNNaffinity) | 0.557 | 0.555 | 0.024 | 0.018 | 0.86 | 0.66 | 0.94 | 0.71 | 1.13 | 1.13 |
 | ||
| Fig. 8 Evaluation of screening performance on the GPCRrecent dataset (N = 16). (A–C) Boxplots comparing the distributions of (A) AUROC, (B) BEDROC (a = 80.5) and (C) enrichment factors at thresholds of 0.5%, 1.0%, and 5.0%, across different screening methods. (D and E) Impact of ligand similarity on the screening performance indicated by BEDROC scores. For each active compound in the GPCRrecent dataset, ECFP4 fingerprints were computed for (D) the entire molecule or (E) the Murcko scaffold. The minimum Tanimoto similarity to all ligands in the RCSB PDB was then determined. Data point represents the mean value across targets for each metric. Targets for which no active compounds met the specified similarity threshold were excluded from metric calculations. | ||
When taking ligand similarity into account (Fig. 8D and E), downward trends persist in the BEDROC metrics across all methods, but the overall decline does not substantially alter their relative rankings. AF3-like approaches, when used with their inherent confidence metrics, continue to substantially outperform other approaches in mean BEDROC, reinforcing their robustness in early enrichment despite dataset variations.
Performance on a subset of LIT-PCBA dataset
One reviewer noted that the negative samples in both the DEKOIS2.0 and GPCRrecent datasets are artificially-generated decoys, which might introduce potential bias. In contrast, LIT-PCBA, another widely-accepted benchmark for VS, contains active and inactive compounds derived from real-world screening campaigns. We therefore evaluated Protenix and Boltz-2 on this dataset, with Glide and Gnina as baselines. Of note, due to the significant computational cost associated with co-folding approaches, our evaluation was limited to a subset of five targets, each containing fewer than 100000 compounds. Furthermore, given the substantial variation in the ratio of actives to inactives across different targets in this benchmark (Table S2), we employed not only the conventional EF1% but also the normalized enrichment factor (NEF).49 The NEF accounts for disparities in the active-inactive ratio, thereby allowing a more direct and fair comparison of model performance across targets.
As shown in Table 4, performance varies considerably across targets: Boltz-2 (ipTM) and Boltz-2 (min-iPAE) each achieve top performance on two targets, while Protenix (ipTM) and Gnina (CNNaffinity) each lead on one. When taking the average NEF1% into account, Boltz-2 (min-iPAE) performs the best, followed by Boltz-2 (ipTM) and Protenix (ipTM), all of which still demonstrate overall superiority over the approaches implemented in Glide and Gnina. We further analyzed the influence of ligand similarity on screening performance (Fig. S14). But unfortunately, this analysis did not yield clear trends as both EF and NEF are highly susceptible to the number of active compounds, and the limited number of targets also prevents drawing statistically consistent conclusions. Despite so, AF3-like approaches show encouraging performance in this straightforward evaluation, further corroborating the findings from the previous datasets.
000 compounds each
| Method | ESR_ago | ESR_antago | PPARG | TP53 | MAPK1 | Average | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EF1% | NEF1%a | EF1% | NEF1% | EF1% | NEF1% | EF1% | NEF1% | EF1% | NEF1% | EF1% | NEF1% | |
| a The normalized enrichment factor (NEFx%) is calculated by dividing the observed EFx% by its theoretical maximum (EFmax) at a threshold x%. This normalization confines NEF to a [0, 1] range, which corrects for disparities in the ratio of actives to inactives among different targets and makes the results directly comparable. | ||||||||||||
| Boltz-2 (ipTM) | 7.69 | 0.077 | 3.88 | 0.078 | 25.62 | 0.259 | 0.00 | 0.000 | 3.24 | 0.032 | 8.09 | 0.089 |
| Boltz-2 (min-iPAE) | 15.37 | 0.154 | 2.91 | 0.059 | 25.62 | 0.259 | 0.00 | 0.000 | 2.59 | 0.026 | 9.30 | 0.100 |
| Protenix (ipTM) | 7.69 | 0.077 | 1.94 | 0.039 | 21.96 | 0.222 | 5.00 | 0.093 | 1.30 | 0.013 | 7.58 | 0.089 |
| Glide_SP | 7.69 | 0.077 | 1.94 | 0.039 | 18.30 | 0.185 | 3.75 | 0.070 | 2.27 | 0.023 | 6.79 | 0.079 |
| Gnina (Vina) | 7.69 | 0.077 | 3.88 | 0.078 | 0.00 | 0.000 | 1.25 | 0.023 | 1.30 | 0.013 | 2.82 | 0.038 |
| Gnina (CNNscore) | 7.69 | 0.077 | 2.91 | 0.059 | 18.30 | 0.185 | 0.00 | 0.000 | 1.62 | 0.016 | 6.10 | 0.067 |
| Gnina (CNNaffinity) | 7.69 | 0.077 | 4.85 | 0.098 | 0.00 | 0.000 | 1.25 | 0.023 | 1.95 | 0.019 | 3.15 | 0.044 |
Structural analysis of top-ranking hit compounds
Although AF3-like approaches show certain advantages in enrichment metrics, it remains unclear whether they could identify structurally distinct active compounds compared to conventional methods. Taking the GPCRrecent dataset as an example, we first investigated whether different approaches exhibit distinct ranking preferences. Fig. S15 presents the pairwise Spearman's rank correlation coefficients between screening tools for active compounds (Fig. S15A) and all compounds (Fig. S15B). As expected, ipTM and min-iPAE combined with Boltz-2 predictions demonstrate high correlations (most values exceeding 0.8) across all the targets, which can be attributed to their highly similar ranking mechanisms. Classical scoring functions, whether used with their original docking engines or Protenix-predicted structures, exhibit moderate correlations for most targets. In contrast, correlations for other groups, even including Boltz-2 (ipTM) and Protenix (ipTM), are generally lower, indicating divergent ranking behaviors.We further analyzed the active compounds enriched in top-100 and top-500 rankings. As shown in Fig. 9, the overlap rates between methods are largely consistent with the ranking correlations. The distinct ranking preferences directly explain the generally low overlap rates observed between most method pairs. Notably, some active compounds in certain targets (e.g., O15552, P13945, Q5NUL3 and Q8TDV5) seem to be easily enriched by almost all the approaches, thus leading to relatively high overlaps across pairs. But for others, the overlaps are more moderate, particularly in the top-100 range. Among all pairs, the actives identified by Boltz-2 (ipTM) and Boltz-2 (min-iPAE) consistently show high overlap, but when paired with other approaches, the metric remarkably decreases. These trends could also be observed in analyses based on average molecular similarities (Fig. S16) and scaffold similarities (Fig. S17).
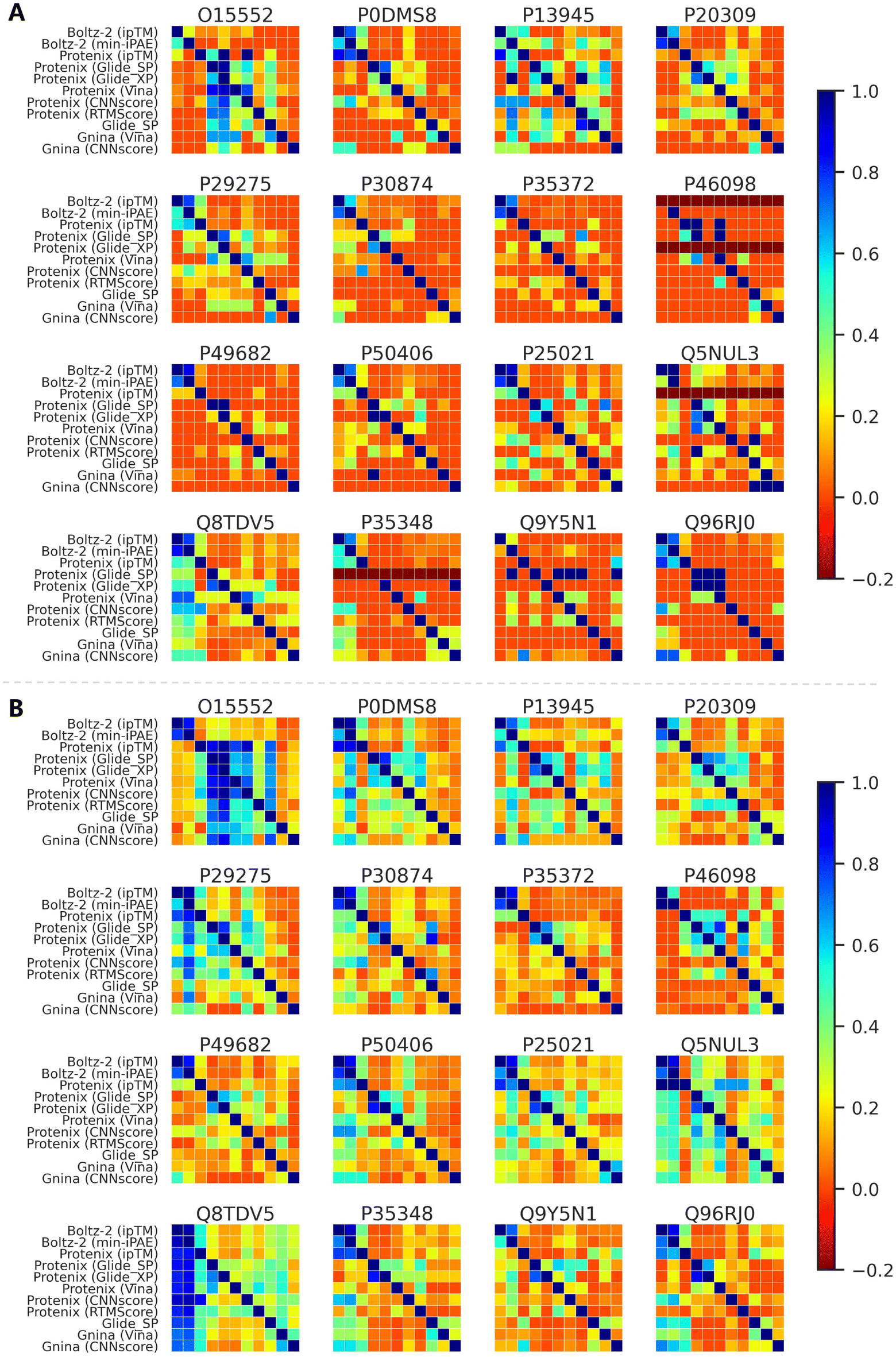 | ||
| Fig. 9 Overlap rates of active compounds among (A) top 100 and (B) top 500 compounds identified by different screening tools across targets in GPCRrecent dataset. An active compound identified by both tools in a comparison is considered an overlap. The overlap rate for a row method is calculated as the percentage of its active compounds that are also found by each column method. A value below 0 indicates that the row method did not identify any active compounds at the specified cutoff. | ||
In summary, while high overlap or structural similarity among actives enriched by different tools does occur for some targets, this appears to stem from the inherent properties of those targets or datasets, rather than from methodological biases. Overall, this analysis confirms that screening methods with distinct ranking mechanisms are capable of enriching structurally diverse compounds. These findings also underscore the value of employing multiple screening strategies in practical VS projects to identify the compounds with novel scaffolds.
Assessment of computational efficiency
The substantial computational cost of AF3-like methods may pose a significant constraint on their practical application. To investigate this further, we simply benchmarked the three co-folding methods involved in this study on five representative targets from the DEKOIS2.0 dataset, each with 400 randomly selected decoys. All experiments were performed on a single NVIDIA H100 GPU with 80 GB of memory. For each target, pre-computed MSAs and ligand structures in SMILES format were fed as input, and computational efficiency was measured by averaging the total time per compound.As summarized in Table 5, AF3 takes approximately 18.42 s to obtain 5 predictions for a given protein–ligand pair, while Protenix requires 34.58 s. When the number of samples is reduced to 1, the corresponding times decrease to 12.99 and 12.38 s, respectively. The latest Boltz-2 appears to be the fastest, taking about 17.77 or 7.79 s per run depending on whether the affinity module is enabled. It should be noted that runtimes vary substantially across different targets, influenced by factors such as token length, number of atoms and MSA depth. Additionally, modeling complexes involving entities beyond a single protein chain and ligand (e.g., additional protein chains or ligands) may introduce extra computational overhead.
| Method | Number of samples | a2a | ar | cdk2 | ctsk | hdac2 | Average |
|---|---|---|---|---|---|---|---|
| AF3 | 5 | 26.34 | 15.36 | 17.48 | 10.28 | 22.63 | 18.42 |
| 1 | 19.14 | 10.64 | 12.82 | 6.70 | 15.66 | 12.99 | |
| Protenix | 5 | 45.83 | 29.70 | 31.80 | 30.01 | 35.56 | 34.58 |
| 1 | 20.09 | 9.46 | 10.47 | 8.49 | 13.41 | 12.38 | |
| Boltz-2 | 1 | 21.48 | 15.43 | 16.83 | 16.17 | 18.93 | 17.77 |
| Boltz-2 (without affinity module) | 1 | 10.83 | 6.14 | 7.11 | 6.05 | 8.79 | 7.79 |
Beyond inference time, two other computational factors also warrant consideration. First, although MSA generation could be performed once per protein target in a typical VS campaign, the time required differs markedly among tools: AF3, using the recommended HMMER-based workflow, takes 20–30 minutes, whereas the MMseqs2-based implementation in Protenix and Boltz-2 completes in under 1 minute. Second, AF3 is substantially more memory-intensive than the other two methods. During inference in our tests, AF3 consistently occupies about 60 GB of GPU memory and could fail on hardware with limited resources. In contrast, both Protenix and Boltz-2 maintain low GPU memory usage (∼5 GB), representing a significant improvement over the original AF3 framework in this regard.
Therefore, compared to traditional docking tools27,29,31 that typically require seconds to tens of seconds per compound on a single CPU core, and early AI-based docking tools like KarmaDock34 that operate at the millisecond level, current AF3-like approaches do not hold an advantage in terms of computational efficiency, particularly given the scarcity of high-performance GPUs. However, they may still serve as valuable components in large-scale VS pipelines, acting as refinement tools for further enriching screening libraries.
Pose examination
The reliability of predicted binding modes is also a critical consideration in structure-based approaches. Buttenschoen et al.50 introduced the “PB-validity” metric to assess the physical plausibility of binding poses, revealing that early AI-based docking methods struggled to generate physically valid conformations. While AF3 maintains robust performance in pose reconstruction when taking “PB-validity” into consideration,2 its generalizability to chemical space beyond crystalized entities remains unexplored.To assess this further, we examined the binding poses generated by three co-folding approaches on DEKOIS2.0 dataset. As outlined in Table 6, despite multiple strategies implemented in AF3 to minimize structural clashes, certain failure modes persist. Across all predicted poses (five per protein–ligand pair), 0.273% (AF3), 0.432% (Protenix), and 0.327% (Boltz-2) of cases exhibit severe structural anomalies, rendering them incompatible with automated processing via the Protein Preparation Wizard51 module of Schrödinger. Notably, these issues were more frequent in decoys than in active compounds (0.280% vs. 0.070% for AF3, 0.445% vs. 0.057% for Protenix, and 0.333% vs. 0.158% for Boltz-2), suggesting their reduced applicability to diverse chemical space.
| Pose generator | Number of poses | Metric | Poses failing to be processed by Protein Preparation Wizard (actives/decoys/total) | Poses considered as ‘PB-invalid’ using PoseBusters toolkit (actives/decoys/total) |
|---|---|---|---|---|
| AF3 | Five | Number | 11/1325/1336 | 155/12218/12373 |
| Ratio | 0.070%/0.280%/0.273% | 0.982%/2.586%/2.534% | ||
| Top-ranked | Number | 0/200/200 | 30/2400/2430 | |
| Ratio | 0%/0.204%/0.211% | 0.950%/2.538%/2.487% | ||
| Protenix | Five | Number | 9/2106/2115 | 124/8537/8661 |
| Ratio | 0.057%/0.445%/0.432% | 0.787%/1.811%/1.778% | ||
| Top-ranked | Number | 3/467/470 | 25/1710/1735 | |
| Ratio | 0.095%/0.493%/0.480% | 0.793%/1.815%/1.782% | ||
| Boltz-2 | One | Number | 5/315/320 | 26/1994/2020 |
| Ratio | 0.158%/0.333%/0.327% | 0.825%/2.112%/2.070% |
Fig. 10 illustrates some representative structural defects, which primarily arises from the unrealistic predicted atomic distances in certain regions. These include distorted aromatic rings (Fig. 10A–D), spurious macrocycles resulting from unnaturally close halogen interactions in distal rings (Fig. 10E and F), and incorrect placement of uncommon functional groups such as adamantane (Fig. 10G), trifluoromethyl (Fig. 10H) and phosphinimine moieties (Fig. 10I). While some errors could be easily corrected through manual intervention, many remain intractable due to severe structural distortions.
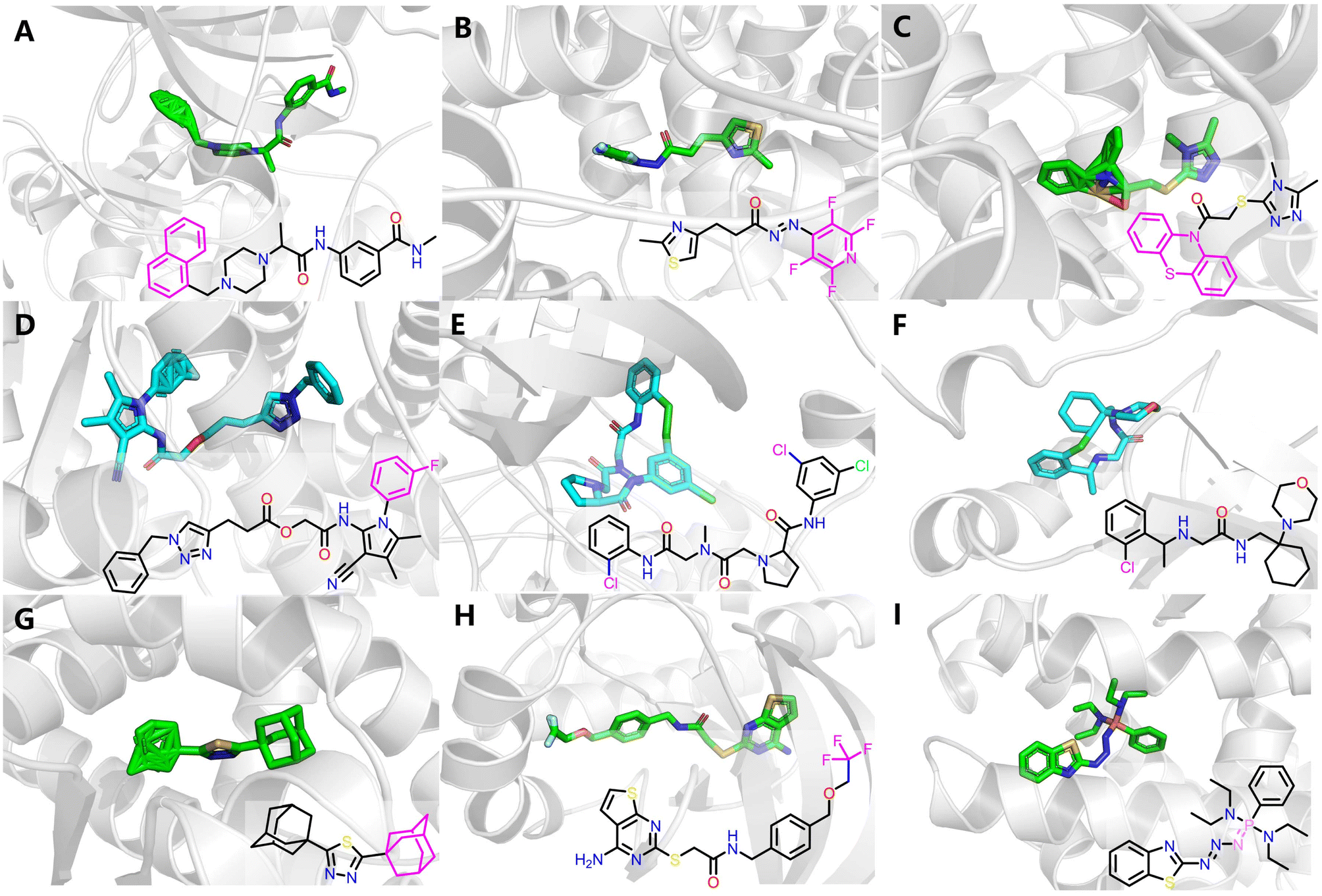 | ||
| Fig. 10 Some representative poses that fail to produce physically valid structures by AF3, including (A–D) distorted aromatic rings, (E and F) spurious macrocycles resulting from unnaturally close halogen interactions in distal rings, and (G–I) incorrect placement of uncommon functional groups. The substructures that have significant structural distortions are colored in magenta. | ||
The incorporation of a clash penalty term in AF3's Ranking score substantially mitigates such failures (e.g., error ratio decreases from 0.273% to 0.211% when using top-ranked poses), whereas Protenix even shows a slight increase (0.432% vs. 0.480%), potentially due to insufficient weighting of steric clashes in its inherent confidence scoring. Following dedicated pose preparation, 2.534% (AF3), 1.778% (Protenix) and 2.070% (Boltz-2) of poses still fail the PoseBusters test, indicating residual minor implausibility. Despite so, these failures are predominantly superficial, exerting minimal impact on subsequent protein–ligand interaction analysis. In a real-world VS scenario, should such failures occur among top-ranked hits, they could likely be addressed through manual correction or simply discarded.
Conclusions
In this study, we conducted a systematic assessment of how AF3-like approaches could be applicable to structure-based VS, using Protenix, AF3 and Boltz-2 as representative models. Our assessment commenced with routine investigation on the well-established DEKOIS2.0 datasets, where AF3 demonstrated exceptional screening performance by relying solely on its intrinsic confidence scores such as min-iPAE and ipTM for compound raking. While the incorporation of third-party scoring schemes failed to enhance screening efficacy, it did confirm both AF3 and Protenix as reliable pose generators. Further analysis revealed performance degradation in three extreme scenarios: (1) when chemically similar active ligands were gradually excluded from test sets, (2) when evaluated on our newly curated dataset consisting of GPCR targets whose structures were rarely represented in model training, and (3) when assessed on a subset of LIT-PCBA dataset where true inactives were incorporated as negative samples. Notably, despite these challenges, these AF3-like approaches consistently maintained superior performance compared to conventional docking tools. Beyond enrichment-based metrics, we also investigated the structural novelty among top-ranking actives identified by different methods, confirming that screening approaches with distinct ranking mechanisms can enrich structurally diverse compounds. Finally, evaluation of the binding poses predicted by three AF3-like apporaches showed that the majority exhibited physically plausible conformations, although some structural anomalies were still observed.Despite these promising observations, it is important to acknowledge that current AF3-like approaches still face significant computational challenges, as evidenced by a preliminary efficiency assessment in this study. Substantial optimizations will be still required to enable their scalable application in high-throughput VS of ultra-large chemical libraries without compromising performance. On the other hand, recent developments like Boltz-2 have sought to unify structure and affinity prediction within a foundation model, relying primarily on affinity prediction model trained from massive affinity data for final compound ranking. Although this approach has shown impressive results, its heavy dependence on affinity data may compromise generalizability to external datasets, an aspect that could not be thoroughly evaluated in the present study and warrants further investigation. In contrast, AF3's structure-based ranking paradigm, relying on confidence scores derived from predicted complex quality, appears to offer a robust and generalizable solution for structure-based VS. It should be noted, however, that our benchmarking, like any retrospective study, relies on the availability of known active ligands, and thus does not involve entirely novel targets. The practical utility of AF3-like approaches ultimately requires validation through prospective experimental studies.
Notwithstanding these considerations, our study underscores the significant potential of AF3-like approaches in structure-based VS, demonstrating both excellent enrichment capabilities through their intrinsic confidence metrics and reliable pose generation. We expect this paper may provide valuable insights for applying AF3-derived methods in this post-AF3 era.
Methods
Datasets
Three complementary datasets, namely DEKOIS2.0,25 GPCRrecent and LIT-PCBA26 were involved in this study for assessing the VS performance of AF3-like approaches. DEKOIS2.0, a well-established benchmark dataset widely used for evaluating both physics-based and AI-powered docking tools,34–36,38,39,52 served as our primary reference. The dataset originally consists of a total of 81 structurally diverse targets, each containing 40 experimentally-validated active compounds from BindingDB42 and 1200 property-matched decoys selected from ZINC.53 Notably, two targets (pygl-in and pygl-out) were excluded in this study since their protein–protein interface binding characteristics make structural modeling computationally expensive for current AF3-like methods, thus resulting in a final set of 79 targets for evaluation.The GPCRrecent dataset was additionally curated here due to the fact that DEKOIS2.0 had been released over ten years and its template protein structures might exhibit a significant overlap with AF3's training data. This complementary set exclusively contains targets whose first crystal structures were determined after 2022, thereby guaranteeing complete temporal separation from any AF3 training samples. The dataset construction workflow is depicted in Fig. S1. Specifically, we first queried the Uniprot54 and BindingDB for targets meeting two criteria: (1) its first crystal structure was released after 2022, and (2) corresponding ligands were available in BindingDB. This yielded 47 eligible UniProt entries, predominantly G protein-coupled receptors (GPCRs). This bias arises because GPCRs exhibit two key characteristics: their crystal structures are historically challenging to resolve with most determinations only becoming possible recently through the wide spread of cryo-electron microscopy (cryo-EM), while their ligand chemical spaces have been extensively explored despite previous structural limitations. Given these considerations, we specifically focused our analysis on these GPCR targets. Active ligands were collected from BindingDB by selecting compounds with their activity (IC50, EC50, Ki, or Kd) values below 10 µM while excluding peptide-like inhibitors and natural products with molecular weights exceeding 600. Notably, unlike conventional practices that specifically excluded the compounds with EC50 annotations, we retained these entries as they usually provided functional insights into agonist/antagonist properties. Following standardization using RDKit toolkit,55 the active compounds were clustered based on their Murcko scaffolds,48 with only the most potent representative per cluster retained. For targets with excessive actives, a maximum of 300 diverse scaffolds were selected based on ECFP4 fingerprints,47 and targets with fewer than 200 actives were also removed, ultimately resulting in 16 GPCRs. Decoys were generated using a streamlined implementation of TocoDecoy56 at a 1:50 active-to-decoy ratio. Compared to DEKOIS2.0 that sources decoys from existing libraries, TocoDecoy employs a conditional recurrent neural network (cRNN) to synthesize property-matched decoys, enabling broader chemical space coverage. The detailed composition of the dataset could be found in Table S1 and Fig. S1.
LIT-PCBA, another widely-accepted VS benchmark, is characterized by experimentally verified bioactivities for all compounds. The full set contains a total of 15 targets, 10030 confirmed actives and 2798737 confirmed inactives. However, due to the high computational cost of those co-folding approaches, only a subset of five targets (Table S2), each with fewer than 100000 compounds, were involved in this study. Notably, MTORC1 was excluded despite meeting the size criterion due to ambiguous binding sites in the recommended PDB structures.
While AF3-like approaches feed only one-dimensional sequences for both proteins and ligands as inputs, some structure-based baseline methods employed in this study require 3D structural information. For the DEKOIS2.0 dataset, we used the provided well-established protein templates and initial ligand conformers, but for LITPCBA, only a single structure was selected from multiple recommended PDB entries (Table S2) to perform baseline docking calculations. For GPCRrecent dataset, we simply retrieved the lowest-resolution protein structure for each UniProt entry from the PDB database and performed comprehensive structural preparation using Protein Preparation Wizard51 module in Schrödinger (version 2020-4). This preparation included the removal of redundant chains and water molecules, assignment of bond orders, addition of hydrogen atoms, completion of missing side chains, optimization of hydrogen-bond networks, and system minimization using the OPLS-2005 (ref. 57) force field until the root-mean-square deviation of heavy atoms averaged at 0.30 Å. The protonation states of residues at pH 7.0 were determined using PROPKA 58while those of co-crystallized ligands using Epik.59 For ligands in both the GPCRrecent and LIT-PCBA datasets, we used the LigPrep module with default settings to generate appropriate tautomers, protonation states, stereoisomers, and low-energy 3D conformers.
Complex structure prediction
Three deep learning-based structure prediction tools, i.e., Protenix (version 0.4.4),13 AF3 (version 3.0.0)2 and Boltz-2 (version 2.0.0),14 were explored in this study. For all these approaches, truncated protein sequences derived from corresponding PDB entries were employed to generate multiple sequence alignments (MSAs) beforehand. Specifically, AF3 employed the Jackhmmer60 module in HMMER61 suite (version 3.4.0) for MSA construction, while Protenix and Boltz-2 utilized the MMseqs2 (ref. 62) web service, all following official recommendations with default settings. To enhance computational efficiency in high-throughput VS, we optimized the inference pipelines of these approaches before conducting individual structure predictions. For AF3 and Protenix, five predictions were generated using a fixed random seed (seed = 1), whereas for the supplemented Boltz-2, the recommended single model under the same random seed were produced to conduct both structure prediction and binding affinity prediction. All other parameters remained at their default or recommended values.Scoring schemes
These AF3-like approaches could output multiple confidence metrics to estimate the reliability of predicted structures, and some of these metrics have shown great promise in capturing crucial interactions between multiple chains. Below are the scoring schemes explored in this study:In addition to utilizing these built-in scoring metrics for compound ranking, we further investigated several third-party rescoring methods, including well-recognized physics-based approaches (Glide27,65 and AutoDock Vina28), and recently-emerged AI-based scoring functions (Gnina,32 RTMScore,38 PIGNet2,44 PLANET,43 and IGModel45) with varying mechanisms. The implementation details for each approach was set default or recommended, unless otherwise described below, unless otherwise described below.
Of note, both AF3 and Protenix output structures in CIF format, which lacks bond information and may not be compatible with certain third-party rescoring methods. Hence, we preprocessed all predicted structures using the Protein Preparation Wizard module, and for any structure that remained unrecognizable, we assigned them an extremely low score. For each protein–ligand pair, five predictions could be obtained through structure prediction. These structures could then be re-ranked using the selected rescoring schemes. Alternatively, they could first be ranked by the built-in confidence scores to select the most reliable structure, followed by application of the specific rescoring scheme for VS.
Baseline docking results
For DEKOIS2.0 dataset, we primarily utilized previously published docking results34–36,38,39 for baseline comparison, while for GPCRrecent dataset and LIT-PCBA subset, we performed new docking calculations using both Glide SP and Gnina. In both cases, the binding box was defined based on the crystallized ligands, with remaining parameters maintained at default settings or as specified earlier. For Glide, the Glide SP score was employed as the primary metric to rank the compounds, while for Gnina, three distinct scoring metrics, i.e., Vina score, CNNscore, and CNNaffinity, were individually investigated.Evaluation metrics
Consistent with prior studies,38,39,52,69 we assessed VS performance primarily based on three well-recognized metrics, including the area under the receiver operating characteristic curve (AUROC),70 Boltzmann enhanced discrimination of receiver operating characteristic (BEDROC, a = 80.5),71 and enrichment factors (EFs). The ROC curve, which plots the true positive rate against the false positive rate, provides a robust measure of overall model performance. The corresponding AUROC value ranges from 0 (complete failure) to 0.5 (random prediction) to 1 (perfect classification). BEDROC incorporates a weighting parameter α into AUROC to emphasize early recognition capability. Here, we set α = 80.5, meaning that the top 2% of ranked molecules contributed to 80% of the BEDROC score. EFx%, defined as the ratio of true positives found in the top x% of ranked compounds relative to random selection, offers a more intuitive measure of early recognition. We evaluated EF at 0.5%, 1%, and 5% thresholds to capture performance across different screening depths. For LIT-PCBA where the ratio of actives to inactives across different targets varies substantially across targets, we supplemented EF1% with the normalized enrichment factor (NEF).49 The NEF is calculated by dividing the observed EFx% by its theoretical maximum, enabling a direct and fair cross-target performance comparison.The metrics for evaluating hit similarity were adapted from a previous study72 that systematically analyzed molecular and scaffold similarities among actives identified by different screening methods within the top-100 and top-500 ranked compounds. Murcko scaffolds48 were generated using the GetScaffoldForMol method from the MurckoScaffold module in RDKit,55 and structural similarities were quantified using Tanimoto coefficients derived from ECFP4 fingerprints.47
The physical plausibility of binding poses was evaluated using the PoseBusters50 toolkit. Since ground-truth reference poses were unavailable for direct comparison, our assessment relied on 14 out of 18 checks that operated independently of known ligand structures.
Author contributions
TJ and CS designed the research study and wrote the paper; CS, XZ, SG, OZ, QW, GD, YZ and LJ performed the experiments and data analysis; PP, YK, QZ and CH helped interpret the results with constructive discussions. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The DEKOIS2.0, GPCRrecent, LIT-PCBA datasets are available at http://www.dekois.com, https://zenodo.org/records/16826965 and https://drugdesign.unistra.fr/LIT-PCBA, respectively. The source codes and execution details of AF3, Protenix and Boltz-2 can be found at https://github.com/google-deepmind/alphafold3, https://github.com/bytedance/Protenix and https://github.com/jwohlwend/boltz, respectively. The source data generated and analyzed in this study at https://zenodo.org/records/17568813.Supplementary information (SI): additional dataset details (Tables S1, S2 and Fig. S1 and S2) and extended results (Fig. S3–S17) that support this study. See DOI: https://doi.org/10.1039/d5sc06481c.
Acknowledgements
This study was financially supported by “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2025C01117), National Natural Science Foundation of China (22303081, 22503081), Zhejiang Provincial Natural Science Foundation of China (LMS25B030003) and China Postdoctoral Science Foundation (2024M762886). In addition, the authors sincerely appreciate Prof. Lei Xu from Jiangsu University of Technology for his expert assistance with molecular modeling using Schrödinger software, which was instrumental to this study.References
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Docking and scoring in virtual screening for drug discovery: methods and applications, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS PubMed.
- V. T. Sabe, T. Ntombela, L. A. Jhamba, G. E. Maguire, T. Govender, T. Naicker and H. G. Kruger, Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review, Eur. J. Med. Chem., 2021, 224, 113705 CrossRef CAS PubMed.
- F. Ballante, A. J. Kooistra, S. Kampen, C. de Graaf and J. Carlsson, Structure-based virtual screening for ligands of G protein-coupled receptors: what can molecular docking do for you?, Pharmacol. Rev., 2021, 73, 1698–1736 CrossRef PubMed.
- X. Zhang, C. Shen, H. Zhang, Y. Kang, C.-Y. Hsieh and T. Hou, Advancing Ligand Docking through Deep Learning: Challenges and Prospects in Virtual Screening, Acc. Chem. Res., 2024, 57, 1500–1509 CrossRef CAS PubMed.
- C. Shen, J. Ding, Z. Wang, D. Cao, X. Ding and T. Hou, From machine learning to deep learning: Advances in scoring functions for protein–ligand docking, Wiley Interdiscip. Rev.-Comput. Mol. Sci., 2020, 10, e1429 CrossRef CAS.
- H. Li, K. H. Sze, G. Lu and P. J. Ballester, Machine-learning scoring functions for structure-based virtual screening, Wiley Interdiscip. Rev.-Comput. Mol. Sci., 2021, 11, e1478 CrossRef.
- M. Fischer, R. G. Coleman, J. S. Fraser and B. K. Shoichet, Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery, Nat. Chem., 2014, 6, 575–583 CrossRef CAS PubMed.
- C. Shen, X. Han, H. Cai, T. Chen, Y. Kang, P. Pan, X. Ji, C.-Y. Hsieh, Y. Deng and T. Hou, Improving the Reliability of Language Model-Predicted Structures as Docking Targets through Geometric Graph Learning, J. Med. Chem., 2025, 68, 1956–1969 CrossRef CAS PubMed.
- C. D. Team, J. Boitreaud, J. Dent, M. McPartlon, J. Meier, V. Reis, A. Rogozhonikov and K. Wu, Chai-1: Decoding the molecular interactions of life, bioRxiv, 2024, preprint, DOI:10.1101/615955.
- J. Wohlwend, G. Corso, S. Passaro, M. Reveiz, K. Leidal, W. Swiderski, T. Portnoi, I. Chinn, J. Silterra, T. Jaakkola and R. Barzilay, Boltz-1 democratizing biomolecular interaction modeling, bioRxiv, 2024, preprint, DOI:10.1101/624167.
- B. A. A. S. Team, X. Chen, Y. Zhang, C. Lu, W. Ma, J. Guan, C. Gong, J. Yang, H. Zhang, K. Zhang, S. Wu, K. Zhou, Y. Yang, Z. Liu, L. Wang, B. Shi, S. Shi and W. Xiao, Protenix-advancing structure prediction through a comprehensive AlphaFold3 reproduction, bioRxiv, 2025, preprint, DOI:10.1101/631967.
- S. Passaro, G. Corso, J. Wohlwend, M. Reveiz, S. Thaler, V. Ram Somnath, N. Getz, T. Portnoi, J. Roy, H. Stark, D. Kwabi-Addo, D. Beaini, T. Jaakkola and R. Barzilay, Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction, bioRxiv, 2025, preprint, DOI:10.1101/659707.
- D. Errington, C. Schneider, C. Bouysset and F. A. Dreyer, Assessing interaction recovery of predicted protein–ligand poses, J. Cheminformatics, 2025, 17, 76 CrossRef CAS PubMed.
- X.-h. He, J.-r. Li, S.-y. Shen and H. E. Xu, AlphaFold3 versus experimental structures: assessment of the accuracy in ligand-bound G protein-coupled receptors, Acta Pharmacol. Sin., 2025, 46, 1111–1122 CrossRef CAS PubMed.
- P. Škrinjar, J. Eberhardt, J. Durairaj and T. Schwede, Have protein–ligand co-folding methods moved beyond memorisation?, bioRxiv, 2025, preprint, DOI:10.1101/636309.
- J. Wee and G.-W. Wei, Evaluation of AlphaFold 3's protein–protein complexes for predicting binding free energy changes upon mutation, J. Chem. Inf. Model., 2024, 64, 6676–6683 CrossRef CAS PubMed.
- F. N. Hitawala and J. J. Gray, What does AlphaFold3 learn about antibody and nanobody docking, and what remains unsolved?, mAbs, 2025, 17, 2545601 CrossRef PubMed.
- S. Zhai, H. Zhao, J. Wang, S. Lin, T. Liu, D. Jiang, H. Liu, Y. Kang, X. Yao and T. Hou, Peppcbench is a comprehensive benchmarking framework for protein–peptide complex structure prediction, J. Chem. Inf. Model., 2025, 65, 8497–8513 CrossRef CAS PubMed.
- F. Zhou, S. Guo, X. Peng, S. Zhang, C. Men, X. Duan, G. Zhu, Z. Wang, W. Li, Y. Mu, L. Zheng, H. L. Liu and S. Wang, Benchmarking AlphaFold3-like Methods for Protein-Peptide Complex Prediction, bioRxiv, 2025, preprint, DOI:10.1101/642277.
- F. Erazo, N. Dunlop, F. Jalalypour and R. Mercado, Predicting PROTAC-mediated ternary complexes with AlphaFold3 and Boltz-1, Digital Discovery, 2025, 4, 3782–3809 RSC.
- Y. Liao, J. Zhu, J. Xie, L. Lai and J. Pei, Benchmarking Cofolding Methods for Molecular Glue Ternary Structure Prediction, J. Chem. Inf. Model., 2025, 65, 11136–11148 CrossRef CAS PubMed.
- Y. Shamir and N. London, State-of-the-art covalent virtual screening with AlphaFold3, bioRxiv, 2025, preprint, DOI:10.1101/642201.
- M. R. Bauer, T. M. Ibrahim, S. M. Vogel and F. M. Boeckler, Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets, J. Chem. Inf. Model., 2013, 53, 1447–1462 CrossRef CAS PubMed.
- V.-K. Tran-Nguyen, C. Jacquemard and D. Rognan, LIT-PCBA: an unbiased data set for machine learning and virtual screening, J. Chem. Inf. Model., 2020, 60, 4263–4273 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- A. N. Jain, Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search, J. Comput.-Aided Mol. Des., 2007, 21, 281–306 CrossRef CAS PubMed.
- N. Zhang and H. Zhao, Enriching screening libraries with bioactive fragment space, Bioorg. Med. Chem. Lett., 2016, 26, 3594–3597 CrossRef CAS PubMed.
- A. T. McNutt, Y. Li, R. Meli, R. Aggarwal and D. R. Koes, GNINA 1.3: the next increment in molecular docking with deep learning, J. Cheminformatics, 2025, 17, 28 CrossRef CAS PubMed.
- W. Lu, Q. Wu, J. Zhang, J. Rao, C. Li and S. Zheng, Tankbind: Trigonometry-aware neural networks for drug-protein binding structure prediction, Adv. Neural Inf. Process. Syst., 2022, 35, 7236–7249 Search PubMed.
- X. Zhang, O. Zhang, C. Shen, W. Qu, S. Chen, H. Cao, Y. Kang, Z. Wang, E. Wang, J. Zhang, Y. Deng, P. Pan, Y. Kang, C.-Y. Hsieh and T. Hou, Efficient and accurate large library ligand docking with KarmaDock, Nat. Comput. Sci., 2023, 3, 789–804 CrossRef CAS PubMed.
- H. Cai, C. Shen, T. Jian, X. Zhang, T. Chen, X. Han, Z. Yang, W. Dang, C.-Y. Hsieh, Y. Kang, P. Pan, X. Ji, J. S. Song, T. Hou and Y. Deng, CarsiDock: a deep learning paradigm for accurate protein–ligand docking and screening based on large-scale pre-training, Chem. Sci., 2024, 15, 1449–1471 RSC.
- D. Cao, M. Chen, R. Zhang, Z. Wang, M. Huang, J. Yu, X. Jiang, Z. Fan, W. Zhang, H. Zhou, X. Li, Z. Fu, S. Zhang and M. Zheng, SurfDock is a surface-informed diffusion generative model for reliable and accurate protein–ligand complex prediction, Nat. Methods, 2025, 22, 310–322 CrossRef CAS PubMed.
- A. Omidi, M. H. Møller, N. Malhis, J. M. Bui and J. Gsponer, AlphaFold-Multimer accurately captures interactions and dynamics of intrinsically disordered protein regions, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2406407121 CrossRef CAS PubMed.
- C. Shen, X. Zhang, Y. Deng, J. Gao, D. Wang, L. Xu, P. Pan, T. Hou and Y. Kang, Boosting protein–ligand binding pose prediction and virtual screening based on residue–atom distance likelihood potential and graph transformer, J. Med. Chem., 2022, 65, 10691–10706 CrossRef CAS PubMed.
- C. Shen, Y. Hu, Z. Wang, X. Zhang, J. Pang, G. Wang, H. Zhong, L. Xu, D. Cao and T. Hou, Beware of the generic machine learning-based scoring functions in structure-based virtual screening, Brief. Bioinform., 2021, 22, bbaa070 CrossRef PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem 2023 update, Nucleic Acids Res., 2023, 51, D1373–D1380 CrossRef PubMed.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. De Veij, H. Ioannidis, D. M. Lopez, J. F. Mosquera, M. P. Magarinos, N. Bosc, R. Arcila, T. Kizilören, A. Gaulton, A. P. Bento, M. F. Adasme, P. Monecke, G. A. Landrum and A. R. Leach, The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods, Nucleic Acids Res., 2024, 52, D1180–D1192 CrossRef CAS PubMed.
- T. Liu, Y. Lin, X. Wen, R. N. Jorissen and M. K. Gilson, BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities, Nucleic Acids Res., 2007, 35, D198–D201 CrossRef CAS PubMed.
- X. Zhang, H. Gao, H. Wang, Z. Chen, Z. Zhang, X. Chen, Y. Li, Y. Qi and R. Wang, Planet: a multi-objective graph neural network model for protein–ligand binding affinity prediction, J. Chem. Inf. Model., 2023, 64, 2205–2220 CrossRef PubMed.
- S. Moon, S.-Y. Hwang, J. Lim and W. Y. Kim, PIGNet2: a versatile deep learning-based protein–ligand interaction prediction model for binding affinity scoring and virtual screening, Digit. Discov., 2024, 3, 287–299 RSC.
- Z. Wang, S. Wang, Y. Li, J. Guo, Y. Wei, Y. Mu, L. Zheng and W. Li, A new paradigm for applying deep learning to protein–ligand interaction prediction, Brief. Bioinform., 2024, 25, bbae145 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The protein data bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- D. Rogers and M. Hahn, Extended-connectivity fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, The properties of known drugs. 1. Molecular frameworks, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
- S. Liu, M. Alnammi, S. S. Ericksen, A. F. Voter, G. E. Ananiev, J. L. Keck, F. M. Hoffmann, S. A. Wildman and A. Gitter, Practical model selection for prospective virtual screening, J. Chem. Inf. Model., 2018, 59, 282–293 CrossRef PubMed.
- M. Buttenschoen, G. M. Morris and C. M. Deane, PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences, Chem. Sci., 2024, 15, 3130–3139 RSC.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J. Comput.–Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- C. Shen, X. Zhang, C.-Y. Hsieh, Y. Deng, D. Wang, L. Xu, J. Wu, D. Li, Y. Kang, T. Hou and P. Pan, A generalized protein–ligand scoring framework with balanced scoring, docking, ranking and screening powers, Chem. Sci., 2023, 14, 8129–8146 RSC.
- J. J. Irwin and B. K. Shoichet, ZINC− a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed.
- U. Consortium, UniProt: the Universal Protein Knowledgebase in 2025, Nucleic Acids Res., 2025, 53, D609–D617 CrossRef PubMed.
- G. Landrum, RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling, Greg Landrum, 2013, 8, 31 Search PubMed.
- X. Zhang, C. Shen, B. Liao, D. Jiang, J. Wang, Z. Wu, H. Du, T. Wang, W. Huo, L. Xu, D. Cao, C.-Y. Hsieh and T. Hou, TocoDecoy: a new approach to design unbiased datasets for training and benchmarking machine-learning scoring functions, J. Med. Chem., 2022, 65, 7918–7932 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- J. C. Shelley, A. Cholleti, L. L. Frye, J. R. Greenwood, M. R. Timlin and M. Uchimaya, Epik: a software program for pK a prediction and protonation state generation for drug-like molecules, J. Comput.-Aided Mol. Des., 2007, 21, 681–691 CrossRef CAS PubMed.
- L. S. Johnson, S. R. Eddy and E. Portugaly, Hidden Markov model speed heuristic and iterative HMM search procedure, BMC Bioinf., 2010, 11, 431 CrossRef PubMed.
- S. R. Eddy, Accelerated profile HMM searches, PLoS Comput. Biol., 2011, 7, e1002195 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- Y. Zhang and J. Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins, 2004, 57, 702–710 CrossRef CAS PubMed.
- R. Evans, M. O'Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, Protein complex prediction with AlphaFold-Multimer, bioRxiv, 2021, preprint, DOI:10.1101/463034.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- P. A. Ravindranath, S. Forli, D. S. Goodsell, A. J. Olson and M. F. Sanner, AutoDockFR: advances in protein–ligand docking with explicitly specified binding site flexibility, PLoS Comput. Biol., 2015, 11, e1004586 CrossRef PubMed.
- R. Quiroga and M. A. Villarreal, Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening, PLoS One, 2016, 11, e0155183 CrossRef PubMed.
- P. G. Francoeur, T. Masuda, J. Sunseri, A. Jia, R. B. Iovanisci, I. Snyder and D. R. Koes, Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design, J. Chem. Inf. Model., 2020, 60, 4200–4215 CrossRef CAS PubMed.
- S. Gu, C. Shen, X. Zhang, H. Sun, H. Cai, H. Luo, H. Zhao, B. Liu, H. Du, Y. Zhao, C. Fu, S. Zhai, Y. Deng, H. Liu, T. Hou and Y. Kang, Benchmarking AI-powered docking methods from the perspective of virtual screening, Nat. Mach. Intell., 2025, 7, 509–520 CrossRef.
- N. Triballeau, F. Acher, I. Brabet, J.-P. Pin and H.-O. Bertrand, Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4, J. Med. Chem., 2005, 48, 2534–2547 CrossRef CAS PubMed.
- J.-F. Truchon and C. I. Bayly, Evaluating virtual screening methods: Good and bad metrics for the "early recognition" problem, J. Chem. Inf. Model., 2007, 47, 488–508 CrossRef CAS PubMed.
- C. Shen, G. Weng, X. Zhang, E. L.-H. Leung, X. Yao, J. Pang, X. Chai, D. Li, E. Wang, D. Cao and T. Hou, Accuracy or novelty: what can we gain from target-specific machine-learning-based scoring functions in virtual screening?, Brief. Bioinform., 2021, 22, bbaa410 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2026 |