
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Data-driven approach to elucidate the correlation between photocatalytic activity and rate constants from excited states
Ryuga
Kunisada
a,
Manami
Hayashi
 a,
Tabea
Rohlfs
b,
Taiki
Nagano
a,
Koki
Sano
a,
Naoto
Inai
a,
Naoki
Noto
*c,
Takuya
Ogaki
d,
Yasunori
Matsui
d,
Hiroshi
Ikeda
d,
Olga
García Mancheño
b,
Takeshi
Yanai
*ae and
Susumu
Saito
*ac
a,
Tabea
Rohlfs
b,
Taiki
Nagano
a,
Koki
Sano
a,
Naoto
Inai
a,
Naoki
Noto
*c,
Takuya
Ogaki
d,
Yasunori
Matsui
d,
Hiroshi
Ikeda
d,
Olga
García Mancheño
b,
Takeshi
Yanai
*ae and
Susumu
Saito
*ac
aGraduate School of Science, Nagoya University, Nagoya, Aichi 464-8602, Japan. E-mail: yanai.takeshi.e4@f.mail.nagoya-u.ac.jp; saito.susumu.c4@f.mail.nagoya-u.ac.jp
bOrganic Chemistry Institute, University of Münster, Münster 48149, Germany
cIntegrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Nagoya, Aichi 464-8602, Japan. E-mail: noto.naoki.f5@f.mail.nagoya-u.ac.jp
dGraduate School of Engineering, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan
eInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Nagoya, Aichi 464-8602, Japan
First published on 25th November 2025
Abstract
Even though excited-state properties play a crucial role in photocatalysis, directly correlating these with photocatalytic activity remains challenging. Herein, we propose a method to elucidate the correlations between the catalytic activity of organic photosensitizers and the rate constants of various excited-state processes through integrating machine learning (ML), quantum chemical calculations, and chemical experiments. This approach was applied to interpolative predictions of the yield of the nickel/photocatalytic formation of C–O bonds and radical additions to alkenes using various organic photosensitizers with satisfactory accuracy (R2 = 0.83 and 0.77 on the test set, respectively). The calculated rate constants obtained through quantum chemical calculations proved to be comparable or even superior to the experimentally measured excited-state lifetimes as descriptors. SHAP-based visual analysis revealed that the rate constants corresponding to transitions from the T1 state provide significant contributions to the interpolative prediction of photocatalytic activity. Additionally, the non-radiative decay process between the S1 and S0 states helps describe the low catalytic activity of poorly emissive photosensitizers. These findings highlight the potential of the proposed method to provide insights into photocatalytic properties that are difficult to obtain using conventional approaches.
Introduction
The excitation of organic molecules induces intriguing phenomena, including photocatalysis.1–9 However, elucidating the relationship between excited-state properties and the resulting photochemical behavior is highly challenging using conventional experimental approaches.The excited-state lifetimes of photosensitizers are considered to play a crucial role in facilitating energy-transfer and electron-transfer processes with substrates, although a long excited-state lifetime does not necessarily correlate with a higher product yield.10 Therefore, clarifying the relationship between photocatalytic activity and excited-state lifetimes, or related properties such as decay rate constants, is essential to understand the behavior of organic photosensitizers (OPSs), which exhibit diverse excited-state properties. Recent advances in quantum chemical calculations have made the prediction of excited-state characteristics, e.g., first-principles prediction of rate constants for various processes in the excited state, more accessible.11–21 However, to elucidate how the complex excited-state properties influence photocatalytic activity using quantum chemical calculations, further integration with robust tools to decipher these relationships is required.
Machine learning (ML) is increasingly being applied across diverse chemical fields, including organic synthesis.22–27 A common application of ML in this field is the prediction of the product yield and selectivity in order to identify the optimal catalyst and reaction conditions.28–34 In addition to its predictive capabilities, ML is valuable for uncovering correlations between inputs, e.g., the properties of substrates and catalysts, and outputs, e.g., the product yield and selectivity of reactions.35–37 Shapley additive explanations (SHAP), a tool based on game theory that has been developed to enhance the interpretability of ML models, is highly useful in this context.38–44 SHAP enables the quantitative assessment of how individual descriptors contribute to the trends in the overall predicted outputs and to the predictions for individual samples. However, despite its utility, the application of ML to the characterization of photocatalytic properties remains an area with considerable room for further development.40,45–54 In particular, approaches that can correlate essential yet elusive excited-state properties, such as rate constants from excited states, with photocatalytic activity are still underdeveloped.
Here, we propose a data-driven approach to estimate the catalytic activity of OPSs using theoretically simulated rate constants from excited states (Fig. 1), whose effectiveness in ML for photocatalysis remains underexplored. Specifically, rate constants for the radiative (kr(S1 → S0)) and non-radiative (kic(S1 → S0)) processes from the S1 to the S0 state, the intersystem crossing (ISC; kisc(S1 → T1)) and reverse ISC (krisc(T1 → S1)) processes between the S1 and T1 states, and the ISC process from the T1 state to the S0 state (kisc(T1 → S0)) were simulated using a combination of time-dependent density functional theory (TD-DFT) and excited-state-dynamics theory based on the thermal vibration correlation function (TVCF). Descriptor sets incorporating these DFT-based properties were used for the ML-based interpolative prediction of the photocatalytic activity in energy-transfer and photoredox reactions, i.e., nickel-catalyzed C–O bond formation and radical addition. Our protocol demonstrates that the integration of advanced quantum chemical calculations into ML represents a pertinent tool to elucidate how complex excited-state characteristics influence photocatalytic activity, thereby highlighting a potential avenue through which data science can contribute to chemical research.
 | ||
| Fig. 1 Rate constants obtained from quantum chemical calculations for the prediction of photocatalytic activity. | ||
Results and discussion
Experimentally measured and theoretically simulated rate constants of representative OPSs
Recently, Inai and Yanai have reported a photochemical study in which a first-principles method based on TD-DFT and TVCF calculations was used to predict the decay rate constants of organic dyes.55 We applied this method to simulate the rate constants of various excited-state processes for OPSs. We first selected four compounds (OPS1, OPS5, OPS7, and OPS47) from among the 60 OPSs used in this study and compared their experimentally measured rate constants with the theoretically simulated ones. A complete list of all OPSs is provided in the SI (Fig. S2).OPS1, commonly known as 4CzIPN, is an organic compound that exhibits thermally activated delayed fluorescence (TADF).56OPS5 and OPS7 are derivatives of OPS1 featuring 3,6-dimethoxycarbazolyl and diphenylamino groups, respectively, instead of the carbazolyl groups of OPS1. OPS47 also contains diphenylamino groups as electron-donor moieties but differs in having a nitrophenyl group as an electron acceptor. The photophysical properties of OPS1 in toluene, including fluorescence lifetimes (τ) and quantum yield (Φ), have already been reported.56–60 Methoxy-substituted OPS5 exhibits shorter τ values and a much lower Φ than OPS1, while OPS7 has a lower Φ value together with a significantly extended τ. OPS47 has a very low Φ with very short τ values and does not exhibit TADF properties. All these τ and Φ values are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| OPS | τ (Φ)b | ΔEST/eV | k r(S1 → S0)/s−1 | k isc(S1 → T1)/s−1 | k risc(T1 → S1)/s−1c |
| a For details of how these properties were obtained, see the SI. b Luminescence lifetimes (τ) and quantum yields (Φ) were measured in toluene under an argon atmosphere. c The excited-state lifetimes and rate constants of OPS1 were obtained from ref. 57. d The Φ and ΔEST values of OPS1 were obtained from ref. 56 and 58–60. e Experimentally determined values are presented. f Theoretically simulated values are presented. g Theoretically simulated kisc(S1 → T1) and krisc(T1 → S1) values, which were calculated using computational (DFT-based) ΔEST values, are presented. h Theoretically simulated kisc(S1 → T1) and krisc(T1 → S1) values, which were calculated using experimental (measurement-based) ΔEST values, are presented. | |||||
| OPS1 | 14.2 ns, 1.8 µsc (0.94)d | 0.08d,e (0.37)f | 1.7 × 107c,e (2.0 × 107)f | 5.1 × 107c,e (2.6 × 107g, 1.1 × 107h) | 2.7 × 106c,e (5.3 × 10g, 2.7 × 106h) |
| OPS5 | 4.8 ns, 0.6 µs (0.06) | 0.03e (0.39)f | 1.1 × 107e (7.8 × 106)f | 2.0 × 108e (4.1 × 106g, 2.5 × 108h) | 2.9 × 105e (2.6 × 10−1g, 1.6 × 107h) |
| OPS7 | 1.7 ns, 34.4 µs (0.20) | 0.12e (0.70)f | 4.8 × 107e (2.4 × 107)f | 5.5 × 108e (3.3 × 105g, 5.4 × 108h) | 4.7 × 104e (6.8 × 10−5g, 3.7 × 105h) |
| OPS47 | 3.5 ns, 9.9 ns (0.02) | 0.15e (0.42)f | 5.7 × 106e (1.9 × 106)f | 2.1 × 108e (1.7 × 108g, 3.8 × 106h) | 6.2 × 105e (6.0 × 102g, 6.9 × 105h) |

The rate constants of these OPSs were experimentally determined (Table 1). Detailed procedures for estimating the rate constants are described in the SI (Experimentally determined rate constants section). The experimentally measured rate constants of OPS1 have also been reported previously.57OPS5 and OPS7 exhibit higher kisc(S1 → T1) values than OPS1, whereas their krisc(T1 → S1) values are lower. OPS47 shows lower kr(S1 → S0) and higher kisc(S1 → T1) values than OPS1, which might explain its poor luminescence properties.
The theoretically simulated rate constants for these OPSs are also summarized in Table 1. The calculations were conducted using the TD-DFT and TVCF methods described in the previous study.55 The underlying DFT and TD-DFT calculations were carried out at the PCM(toluene)-CAM-B3LYP/6-31G(d) level, and the theoretically simulated kr(S1 → S0) values show good agreement with the experimentally determined values for all four OPSs.
To calculate kisc(S1 → T1) and krisc(T1 → S1), the adiabatic singlet–triplet splitting (ΔEST) values of the OPSs are required. Specifically, the TVCF method sums over vibronic levels under the harmonic approximation and employs ΔEST as the detuning parameter in the phase factor rather than a standalone proxy for the activation. Considering that accurately computing ΔEST values via DFT-based methods remains challenging,61–63 we compared the effects of using the computational (DFT-based) or experimental (measurement-based) ΔEST values on the resulting kisc(S1 → T1) and krisc(T1 → S1) values. The ΔEST values of OPS1, OPS5, OPS7, and OPS47 obtained via quantum chemical calculations (OPS1: 0.37 eV; OPS5: 0.39 eV; OPS7: 0.70 eV; OPS47: 0.42 eV) and experiments (OPS1: 0.08 eV; OPS5: 0.03 eV; OPS7: 0.12 eV; OPS47: 0.15 eV) are provided in Table 1. In addition, theoretically simulated kisc(S1 → T1) and krisc(T1 → S1) values that were refined using the experimental ΔEST are also listed in Table 1.
The relative effectiveness of using the computational or experimental ΔEST value for the calculation of the kisc(S1 → T1) value varied on a case-by-case basis. Both methods provided similar kisc(S1 → T1) values for OPS1 (computational ΔEST: 2.6 × 107 s−1; experimental ΔEST: 1.1 × 107 s−1). The computational ΔEST values resulted in better agreement with the experimentally determined kisc(S1 → T1) for OPS47 (computational ΔEST: 1.7 × 108 s−1; experimental ΔEST: 3.8 × 106 s−1), whereas the opposite effect was observed for OPS5 (computational ΔEST: 4.1 × 106 s−1; experimental ΔEST: 2.5 × 108 s−1) and OPS7 (computational ΔEST: 3.3 × 105 s−1; experimental ΔEST: 5.4 × 108 s−1).
In contrast, the experimental ΔEST values were clearly more effective for simulating krisc(T1 → S1). The fully computation-based approach significantly underestimated the krisc(T1 → S1) values (OPS1: 5.3 × 10 s−1; OPS5: 2.6 × 10−1 s−1; OPS7: 6.8 × 10−5 s−1; OPS47: 6.0 × 102 s−1) compared to the cases where the experimental ΔEST values were used (OPS1: 2.7 × 106 s−1; OPS5: 1.6 × 107 s−1; OPS7: 3.7 × 105 s−1; OPS47: 6.9 × 105 s−1). However, despite this underestimation, the former method captured more accurately the relative magnitudes of OPS1 and OPS5 than the latter, indicating that the use of fully computational values in ML is not always inferior.
For processes involving ISC, using the experimental ΔEST value for the TVCF calculations tended to provide values closer to the experimentally determined rate constants, due to the difficulty of accurately estimating ΔESTvia DFT-based approaches. Meanwhile, for compounds with poor emission properties, experimentally determining the ΔEST value is difficult. In addition, a fully computational approach is more promising in terms of ensuring the future applicability of this framework to compounds that have not yet been synthesized. Alternatively, the prediction accuracy of ΔEST can be significantly improved using higher-level wavefunction-based methods such as SCS-CC2.62,63 However, the computational cost of such methods still remains prohibitively high for the relatively large molecular datasets used for ML. Therefore, we chose to use the ΔEST values obtained from DFT-level calculations, i.e., the computational ΔEST values, in the subsequent ML investigations.
Comparison of the model performance in C–O bond-forming reactions
Although the focus of the present study was on quantifying the relative contributions of the model inputs (OPS properties) to the model output (predicted yield) rather than achieving maximum model accuracy, a certain level of model performance is crucial for meaningful feature interpretation. In our case, the challenge lies in attaining reasonable model performance while using only descriptors that are meaningful from a photochemical perspective. Therefore, we first investigated the impact of the employed descriptors on the accuracy of interpolative predictions for C–O bond-forming reactions using various aryl halides,64–66 which are considered to be promoted mainly by energy transfer (Fig. 2).50,67–69 Our dataset included reactions using four different aryl halides, i.e., 4-bromobenzonitrile (CO-a, CO-b), 4-bromobiphenyl (CO-c), methyl 4-bromo-3-methylbenzoate (CO-d), and 4-chlorobenzonitrile (CO-e). For 4-bromobenzonitrile, data for two distinct reaction times (1.5 h: CO-a; 7.5 h: CO-b) were considered. In our previous study, 60 OPSs were tested for each C–O bond-forming reaction,54 resulting in a dataset consisting of 300 data points. The differences among the OPSs were represented by their DFT-derived properties, and the reaction types (i.e., CO-a–CO-e) were encoded using one-hot encoding. In all the ML investigations, the target variable was the product yield. Since the yield was determined using the employed OPS and the reaction type, the developed models can be regarded as ML models for the estimation of the photocatalytic activity in similar reactions. Other details of the ML setup are summarized in Table 2. | ||
| Fig. 2 C–O bond-forming reactions tested in this study. | ||
|
|
|||
|---|---|---|---|
| Entry | Descriptor set | R 2 | RMSE |
| a R 2 and RMSE scores on the test set were averaged over 10 runs (standard deviations in parentheses). | |||
| 1 | RC | 0.79 (0.05) | 13.1 (1.2) |
| 2 | s_RC | 0.78 (0.04) | 13.5 (1.1) |
| 3 | s_RC + ‘EHOMO, fS1, ΔEST, ΔDM’ | 0.83 (0.04) | 11.8 (1.3) |
| 4 | ‘EHOMO, fS1, ΔEST, ΔDM’ | 0.79 (0.06) | 13.0 (1.4) |
| 5 | LT_t | 0.66 (0.06) | 16.9 (1.9) |
| 6 | LT_d | 0.64 (0.08) | 17.2 (1.4) |
| 7 | LT_t + ‘EHOMO, fS1, ΔEST, ΔDM’ | 0.80 (0.05) | 12.8 (1.5) |
| 8 | LT_d + ‘EHOMO, fS1, ΔEST, ΔDM’ | 0.79 (0.05) | 13.1 (1.2) |

In addition to the three theoretically simulated rate constants mentioned earlier, i.e., kr(S1 → S0), kisc(S1 → T1), and krisc(T1 → S1), we also incorporated kic(S1 → S0) and kisc(T1 → S0). The adiabatic singlet–triplet splitting between the S0 and T1 states was required to calculate kisc(T1 → S0) instead of that between the S1 and T1 states, which was used for the estimation of kisc(S1 → T1) and krisc(T1 → S1). In contrast, the rate constant for the radiative process from the T1 state to the S0 state was not included because phosphorescence is intrinsically weak in OPSs and its contribution to the overall excited-state kinetics is negligible. These five rate constants were used directly to generate a descriptor set comprising five descriptors (referred to as RC). Alternatively, scaled descriptors expressed as ratios among the five rate constants were used to generate another five-descriptor set (denoted as s_RC). The method used to prepare the s_RC descriptor set is summarized in the SI (Computational details for the design of descriptors section). A preliminary investigation identified histogram-based gradient boosting (HGB) as an effective ML model (Table S13). Among the rate constants calculated at different levels of theory, those derived from the PCM(toluene)-CAM-B3LYP/6-31G(d) level provided the best model performance for both RC and s_RC, although the differences were not significant (Table S15). Therefore, descriptors calculated at this level were used in subsequent investigations. The mean R2 scores on the test set were 0.79 for RC and 0.78 for s_RC (Table 2, entries 1 and 2), indicating that reasonable interpolative predictions can be achieved using only the rate-constant information.
Subsequently, we examined whether incorporating other physical properties relevant to the photoreactions could lead to more robust models. The additional descriptors include the HOMO (EHOMO) and LUMO (ELUMO) energy levels, the vertical-excitation (absorption) energy of the lowest singlet (ES1) and triplet (ET1) excited states, the corresponding vertical ΔEST, the oscillator strength of the lowest singlet excitation (fS1), and the differences between the ground- and excited-state dipole moments (ΔDM). These descriptors were calculated at the same PCM(toluene)-CAM-B3LYP/6-31G(d) level. Details of the preparation for these descriptors are provided in the SI (Computational details for the design of descriptors section). We compared the model performance of all 127 combinations of these descriptors in conjunction with RC or s_RC, and identified s_RC, EHOMO, fS1, ΔEST, and ΔDM as the best descriptor set (entry 3; R2 = 0.83, RMSE = 11.8). When the rate constants were excluded from the descriptor set, the model performance was lower than the best case (entry 4; R2 = 0.79, RMSE = 13.0), indicating that combining s_RC with other descriptors leads to improved accuracy.
Furthermore, the effectiveness of using experimentally measured excited-state lifetimes as descriptors was examined. The excited-state lifetimes were measured using transient absorption spectroscopy in toluene or DMSO (referred to as LT_t and LT_d, respectively). When either LT_t or LT_d was used as a standalone descriptor, the model performance significantly decreased (entries 5 and 6; LT_t: R2 = 0.66, RMSE = 16.9; LT_d: R2 = 0.64, RMSE = 17.2). Additionally, when either LT_t or LT_d was combined with EHOMO, fS1, ΔEST, and ΔDM, the resulting scores (entries 7 and 8; R2 = 0.79–0.80, RMSE = 13.1–12.8) did not surpass those obtained using the calculated rate constants.
One major issue with constructing a database of experimentally measured excited-state properties is the difficulty of performing all measurements under identical conditions. For example, while most of the excited-state lifetimes in this study were measured using toluene or DMSO as the solvent, some data points were obtained in other solvents, such as acetonitrile (OPS59, OPS60) or DMF (OPS44, OPS56, OPS59, OPS60), due to solubility and related issues (Table S12). Additionally, unlike theoretically simulated rate constants, experimentally measured excited-state lifetimes represent a combined value that encompasses various excited-state processes. These inconsistencies in measurement conditions and the limited ability to distinguish individual excited-state processes may have contributed to the decreased accuracy observed when using LT_t or LT_d. Furthermore, clarifying each individual kinetic parameter requires considerable experimental effort. Therefore, the theoretically simulated rate constants, which can provide more details regarding the molecular excited states, have superior utility as descriptors as well as greater interpretability (vide infra).
SHAP-based analysis for C–O bond-forming reactions
We next used SHAP to quantify the contributions of descriptors to the interpolative predictions in the case where the best model performance was achieved (all data: R2 = 0.96; test set: R2 = 0.88). In this analysis, we generated a SHAP summary plot (Fig. 3a) to capture the overall trends, and SHAP scatter plots (Fig. 3b) to clarify the contribution of each descriptor. In addition, SHAP waterfall plots were used to visually display the feature contributions of individual OPSs for CO-a (Fig. 2). In this reaction, the experimental yields for OPS1, OPS7, and OPS47 were 62%, 88%, and 0%, respectively. Briefly, a higher absolute SHAP value indicates a greater contribution of the corresponding descriptor, and its signum shows whether the contribution affects the prediction positively or negatively. Additional explanations on how to interpret these plots are provided in the SI (Description of SHAP plots section). Descriptors prefixed with ‘reaction_’ represent the one-hot encoding used to identify reaction types. Additionally, the term ‘feature’ is used synonymously with ‘descriptor’. | ||
| Fig. 3 SHAP (a) summary, (b) scatter, and (c) waterfall plots (for CO-a) for the quantification of feature contributions. (d) OPSs discussed in the SHAP-based analysis. Plots including more detailed information are displayed in Fig. S7. The original plots are included in the GitHub repository (https://github.com/Naoki-Noto/P6-20240509-RK). | ||
The SHAP summary plot revealed that ΔEST and EHOMO are the two most impactful descriptors excluding the one-hot encoding descriptors (Fig. 3a). Highly negative SHAP values for ΔEST were observed for high ΔEST values, whereas lower ΔEST values lead to positive SHAP values (Fig. 3b). A range of moderate EHOMO values resulted in highly positive SHAP values, while those outside this range yielded negative SHAP values (Fig. 3b). In the C–O bond-forming reactions, OPSs that meet these conditions to afford positive SHAP values, such as OPS1 and OPS7 (Fig. 3c), generally exhibit better catalytic activity. In contrast, OPSs with high ΔEST values, e.g., OPS56 and OPS58, those with strong reducing capacity, e.g., OPS38, OPS40, and OPS49, and those with strong oxidizing capacity, e.g., OPS59 and OPS60, tend to furnish low product yields (Table S17). This SHAP-based analysis demonstrates the ability to effectively capture the correlations between physical properties and photocatalytic activity.
Next, we analyzed the contributions of the rate constants from the summary plot (Fig. 3a). As mentioned earlier, these descriptors are expressed as ratios of the five processes and are prefixed with ‘s_’. Among the five descriptors derived from rate constants, the descriptors for two processes, i.e., s_krisc(T1 → S1) and s_kisc(T1 → S0), showed greater contributions than the other three. Given that the triplet state of OPSs is highly likely to be involved in photosensitization,4,6,70,71 it is reasonable to assume that descriptors representing transitions from the T1 state to other states are important. Although the s_krisc(T1 → S1) values tended to be underestimated in the employed descriptor set, the SHAP scatter plots revealed that s_krisc(T1 → S1) values within a certain range tend to result in positive SHAP values, whereas those outside this range produce negative SHAP values (Fig. 3b). Similarly, s_kisc(T1 → S0) values that fell within a specific range also tend to exhibit positive SHAP values (Fig. 3b). The SHAP waterfall plots for OPS1 and OPS7 (Fig. 3c) indicate that while EHOMO, fS1, ΔEST, and ΔDM exhibit relatively similar positive SHAP values, s_krisc(T1 → S1) and s_kisc(T1 → S0) are the primary contributors to distinguishing the catalytic activity of OPS1 and OPS7. The predicted yields were 53.85% for OPS1 and 85.15% for OPS7, demonstrating relatively high accuracy. The SHAP values for s_krisc(T1 → S1) were −1.17 for OPS1 and +6.17 for OPS7, while those for s_kisc(T1 → S0) were −2.69 for OPS1 and +8.13 for OPS7. Thus, based on SHAP, 18.16% of the 31.30% difference in predicted yields between OPS1 and OPS7 can be attributed to these two descriptors derived from rate constants.
Meanwhile, OPS47 is structurally distinct from OPS7 in terms of its acceptor moiety, resulting in a significantly lower quantum yield (OPS1: Φ = 0.94; OPS7: Φ = 0.20; OPS47: Φ = 0.02) and catalytic activity compared to OPS1 and OPS7. The SHAP waterfall plots revealed that ΔEST, EHOMO, fS1, and ΔDM, which provided highly positive SHAP values for OPS1 and OPS7, do not contribute positively to the model output for OPS47 (Fig. 3c). Additionally, the negative SHAP value derived from s_krisc(T1 → S1) (−4.36) for OPS47 contributes to distinguishing the catalytic activity of OPS7 and OPS47. The s_kr(S1 → S0) of OPS47 was 2.7 × 10−4 and its s_kic(S1 → S0) was 0.768, indicating that the former is significantly lower and the latter significantly higher compared to those of OPS1 (s_kr(S1 → S0): 0.431; s_kic(S1 → S0): 9.6 × 10−4) and OPS7 (s_kr(S1 → S0): 0.573; s_kic(S1 → S0): 2.4 × 10−4). Unlike in the case of OPS1 and OPS7, these descriptors have negative SHAP values of −6.04 and −4.10 for OPS47. The low s_kr(S1 → S0) and high s_kic(S1 → S0) observed for OPS47 reflect its poor luminescence properties. The low catalytic activity of the OPSs, for which non-radiative decay pathways unrelated to the transition to the triplet state are favored, is consistent with chemical intuition. The ML-derived outcome, in which the s_kr(S1 → S0) and s_kic(S1 → S0) values of OPS47 negatively impact its output, support this notion.
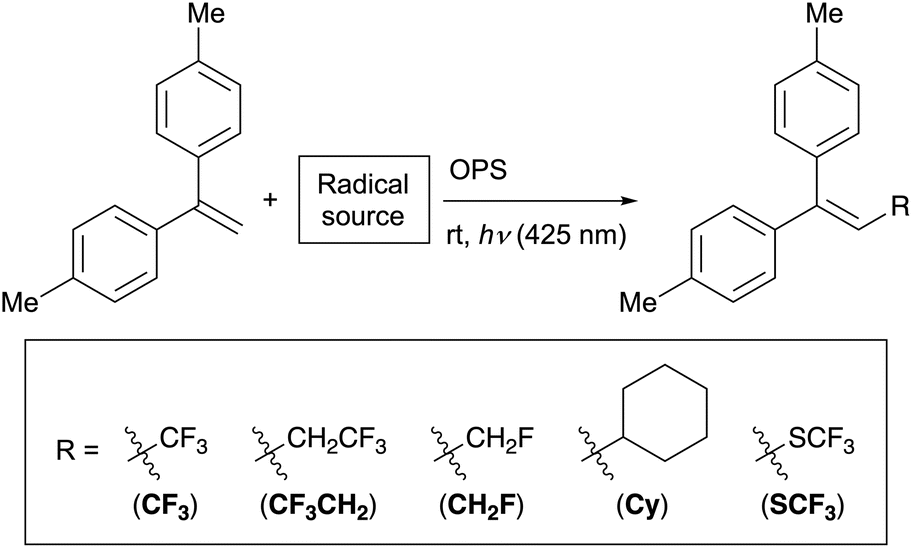
Application to radical–addition reactions to alkenes
We then extended the scope of our ML strategy to photoredox reactions. As target reactions, we selected the addition of trifluoromethyl, 2,2,2-trifluoroethyl, monofluoromethyl, cyclohexyl, and trifluoromethylthio radicals to 1,1-di(p-tolyl)ethylene.72–78 In this study, these reactions to synthesize trisubstituted alkenes are referred to as CF3, CF3CH2, CH2F, Cy, and SCF3, respectively (Table 3). By testing the same database of 60 OPSs for these five photoredox reactions, we constructed a dataset consisting of 300 data points. The HGB-based ML models were constructed using the same setup as that used for the C–O bond-forming reactions (Table 2).|
|
|||
|---|---|---|---|
| Entry | Descriptor set | R 2 | RMSE |
| a For details of reaction conditions, see the ESI. R2 and RMSE scores on the test set were averaged over 10 runs (standard deviations in parentheses). | |||
| 1 | s_RC | 0.67 (0.05) | 19.3 (1.9) |
| 2 | s_RC + ‘ELUMO, fS1, ET1’ | 0.77 (0.03) | 16.1 (1.5) |
| 3 | LT_t + ‘ELUMO, fS1, ET1’ | 0.73 (0.05) | 17.5 (1.6) |
| 4 | LT_d + ‘ELUMO, fS1, ET1’ | 0.75 (0.03) | 16.6 (0.9) |
Since the model performance did not differ significantly among the descriptor sets based on rate constants (Table S16), we continued to use the s_RC descriptor set calculated at the PCM(toluene)-CAM-B3LYP/6-31G(d) level in order to maintain consistency with the results of the C–O bond-forming reactions. Although the model performance using s_RC alone was relatively poor (Table 3, entry 1; R2 = 0.67), combining s_RC with ELUMO, fS1, and ET1 improved the interpolative predictions, yielding a mean R2 score of 0.77 and a mean RMSE of 16.1 on the test set (entry 2). While a long excited-state lifetime is known to contribute to efficient electron transfer, it is not always the sole factor determining the product yield, as reported for similar photoredox reactions.10,79,80 Thus, it is reasonable that the model based solely on s_RC exhibits relatively poor performance, and combining s_RC with other descriptors such as ELUMO improves the model accuracy. When the experimentally obtained LT_t or LT_d descriptor set was used instead of s_RC, the resulting R2 scores were 0.73 and 0.75, respectively (Table 3, entries 3 and 4), confirming the satisfactory performance of an entirely DFT-derived descriptor set.
A SHAP-based analysis was subsequently conducted for the best-performing model (all data: R2 = 0.94; test set: R2 = 0.82). The SHAP bar plot depicting the mean SHAP values of the descriptors indicated that ELUMO, which exhibits a high correlation coefficient with EHOMO (0.86) and is associated with the redox capacity of the OPSs, shows the largest contribution excluding the one-hot encoding descriptors (Fig. 4a). The SHAP scatter plot (Fig. 4b) revealed that a high ELUMO contributes to positive SHAP values, but when the ELUMO is too high, the SHAP values shift negatively. Both oxidizing and reducing properties are important in photoredox reactions, and an excessively high ELUMO values implies a weak oxidizing capacity for an OPS. Therefore, the SHAP-derived result that an excessively high ELUMO negatively influences the product yield appears reasonable. Furthermore, it is known that while the product yield in photoredox reactions is strongly affected by the lifetime of radicals generated in situ, an excessively high ELUMO can negatively influence the lifetime of radicals derived from OPSs and consequently reduce the product yield.81,82 This insight is also consistent with the result obtained from SHAP.
 | ||
| Fig. 4 SHAP (a) bar and (b) scatter plots as well as (c) waterfall plots (for Cy) for the quantification of feature contributions. Waterfall plots including the more detailed information are displayed in Fig. S8. The original plots are included in the GitHub repository (https://github.com/Naoki-Noto/P6-20240509-RK). | ||
The trends in the feature contribution of rate constants in the ML model for the radical–addition reactions are similar to those observed in the ML model for the C–O bond-forming reactions (Fig. 3a and 4a). Notably, s_krisc(T1 → S1) and s_kisc(T1 → S0), which are key descriptors for the C–O bond-forming reactions, also play an important role in the ML model for the radical–addition reactions. Next, SHAP waterfall plots were generated for OPS1 and OPS5 in Cy (Fig. 4c). In this reaction, OPS1 and OPS5 afforded experimental yields of 45% and 0%, respectively. OPS1 and OPS5 differ in the absence or presence of methoxy groups on their skeletons. The predicted yields for OPS1 and OPS5 were 44.11% and 2.52%, respectively, indicating that the ML model can distinguish the difference in product yields based on their physical properties. The SHAP waterfall plots (Fig. 4c) revealed that, while ELUMO exhibits a highly negative SHAP value in both cases (OPS1: −12.87; OPS5: −15.01), ET1 exhibits significantly different SHAP values (OPS1: +3.17; OPS5: −4.24). The s_kic(S1 → S0) and s_krisc(T1 → S1) parameters exhibit significantly more negative SHAP values for OPS5 than OPS1, with larger differences in SHAP values (17.17 and 11.33) than ET1 (7.41). Thus, based on SHAP, 28.50% of the 41.59% difference in predicted yield can be attributed to s_kic(S1 → S0) and s_krisc(T1 → S1).
The higher s_kic(S1 → S0) value of OPS5 (3.4 × 10−2) than that of OPS1 (9.6 × 10−4) supports its poor luminescence properties. The lower s_krisc(T1 → S1) of OPS5 (2.1 × 10−8) than that of OPS1 (1.2 × 10−6) is consistent with the trend in the experimental values. We have long recognized that the methoxy group is a substituent that specifically impairs luminescence properties and catalytic activity,50,77 although explaining the dramatic effects of “MeO” using more fundamental physical properties is challenging. It is noteworthy that incorporating theoretically simulated rate constants into ML enabled us to capture such small yet specific differences. Moreover, as mentioned earlier, our study clarified that when rate constants are generated using the same computational method, their correlation with photocatalytic activity in energy-transfer and photoredox reactions is similar.
Conclusions
We have developed a data-driven approach to elucidate the correlation between the rate constants from excited states and the catalytic activity of organic photosensitizers (OPSs) by integrating machine learning (ML), quantum chemical calculations, and experiments. The theoretically simulated rate constants provided a model performance that was comparable, or even superior, to that obtained using the experimental excited-state lifetimes. A SHAP-based analysis revealed that the rate constants associated with transitions from the T1 state to other states, as well as those from the S1 state to the S0 state, play a crucial role in determining the photocatalytic activity.Among the effective OPSs reported so far, there have been cases where S1-state contributions were observed,80,83 indicating complexity and diversity in their excited-state behavior. Nevertheless, part of the SHAP-derived outcomes demonstrates that, overall, properties associated with transitions from the T1 state play an important role in governing the photocatalytic activity. Researchers are often influenced by biases derived from a limited set of experimental observations, particularly from compounds they are most familiar with. Therefore, incorporating statistical, data-driven approaches can provide a more comprehensive and objective perspective for catalyst design and mechanistic understanding.
Beyond its utility for capturing general trends across the dataset, SHAP is particularly useful for case-by-case analyses. Through this framework, we successfully elucidated the correlations between excited-state properties influenced by subtle structural variations and the corresponding photocatalytic activity. For example, differences were observed in s_kisc(T1 → S0) and s_krisc(T1 → S1) for OPS1 and OPS7 (carbazolyl vs. diphenylamino groups), in s_kr(S1 → S0) and s_kic(S1 → S0) for OPS7 and OPS47 (nitro vs. cyano groups), and in s_kic(S1 → S0) and s_krisc(T1 → S1) for OPS1 and OPS5 (the presence or absence of methoxy groups), which are consistent with experimental observations and chemical intuitions. Statistical analyses based on our dataset suggest that these factors account for the observed differences in catalytic activity. In particular, the developed descriptors, e.g., s_kic(S1 → S0), successfully captured the characteristics of OPSs with poor luminescent properties. Such case-specific analyses are compatible with the nature of organic chemistry.
Although the DFT-level computational approach introduces some numerical uncertainty particularly in kisc(S1 → T1) and krisc(T1 → S1), the resulting relative relationships are sufficient for our ML framework to capture the overall trends, as substantiated by the improved model performance and the agreement between the SHAP-based analysis and experimental interpretations. We show that excited-state properties, which are often difficult to capture experimentally, can be reasonably related to photocatalytic activity at a feasible computational cost. This provides a chemically meaningful contribution beyond purely data-driven aspects. Meanwhile, to further generalize this strategy, continued experimental and computational efforts to construct databases that encompass a broader range of compounds and that incorporate more accurate photophysical properties are essential. For instance, when incorporating complex photosensitizers based on iridium or ruthenium, which are known to be highly effective, we should consider, for example, ultrafast excited-state processes84 and the radiative rate constant associated with the T1–S0 transition, which are requirements that differ significantly from those of OPSs. Developing rational strategies to integrate such differences will be an important challenge for future research.
Author contributions
This study was primarily conducted by R. K., who performed experiments and machine-learning studies, and by M. H., who processed calculations of the descriptors. T. R., T. N., and K. S. supported the photoreaction experiments, while O. G. M. supervised T. R. The computational method used for calculating rate constants in this study was previously reported by N. I. and T. Y., who also contributed to the descriptor design in this study. The photophysical experiments were carried out under the guidance and discussions led by Y. M., with support from T. O. and supervision by H. I. The overall research was directed by N. N., who was also involved in experiments and machine-learning studies, under the supervision of S. S. The initial manuscript was drafted by N. N. with contributions from R. K., M. H., N. I., T. Y., and Y. M., and was edited by all co-authors.Conflicts of interest
There are no conflicts to declare.Data availability
Example code used within this study and corresponding data are available at our GitHub repository (https://github.com/Naoki-Noto/P6-20240509-RK).The data supporting this article have been included in the supplementary information (SI). Supplementary information: full experimental methods including detailed synthetic procedures and characterization data, computational details, and NMR spectra are compiled. See DOI: https://doi.org/10.1039/d5sc06465a.
Abbreviations
| DFT | Density functional theory |
| HGB | Histogram-based gradient boosting |
| ISC | Intersystem crossing |
| ML | Machine learning |
| OPS | Organic photosensitizer |
| RISC | Reverse intersystem crossing |
| SHAP | Shapley additive explanations |
| TADF | Thermally activated delayed fluorescence |
| TD-DFT | Time-dependent density functional theory |
| TVCF | Thermal vibration correlation function |
Acknowledgements
This research was supported by a JSPS/MEXT Grants-in-aid for Young Scientists (grants 25K18029 and 23K13744 to N. N.) and Transformative Research Areas (A) Digi-TOS, KAKENHI (grants 24H01071 and 22H05356 to N. N., as well as 24H01092 and 22H05377 to H. I.), Specially Promoted Research and Transformative Research Areas (A) Green Catalysis Science, KAKENHI (grants 23H05404 and 23H04904 to S. S.), Scientific Research (S), Transformative Research Areas (A) Pi-Molecular Complexity, and Scientific Research (A), KAKENHI (grants 25H00429, 25H01253, 25H01266, and 24H00449 to T. Y.), as well as International Leading Research, KAKENHI (grant 22K21346 to S. S. and T. Y.). T. Y. is grateful for the CPU time grant from the Research Centre for Computational Science, Okazaki, Japan (25-IMS-C083 and 24-IMS-C082) and for support from MEXT Promotion of Development of a Joint Usage/Research System Project: Coalition of Universities for Research Excellence Program (CURE) (grant JPMXP1323015482). O. G. M. and T. R. gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the IRTG 2678 - Functional π-Systems (pi-Sys) (Münster-Nagoya International Research Training group, GRK 2678-437785492) for generous financial support. M. H. acknowledges the “THERS Make New Standards Program for the Next Generation Researchers” (JST SPRING, grant JPMJSP2125). S. S. also acknowledges JST CREST, Japan (grant JPMJCR22L2) for financial support.Notes and references
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 Search PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 Search PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 Search PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 Search PubMed.
- J. R. Ochola and M. O. Wolf, Org. Biomol. Chem., 2016, 14, 9088–9092 Search PubMed.
- Y. Niu, Q. Peng, C. Deng, X. Gao and Z. Shuai, J. Phys. Chem. A, 2010, 114, 7817–7831 CrossRef CAS PubMed.
- Q. Peng, Y. Niu, Q. Shi, X. Gao and Z. Shuai, J. Chem. Theory Comput., 2013, 9, 1132–1143 CrossRef CAS PubMed.
- Q. Ou, Q. Peng and Z. Shuai, J. Phys. Chem. Lett., 2020, 11, 7790–7797 Search PubMed.
- Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 Search PubMed.
- K. Shizu and H. Kaji, J. Phys. Chem. A, 2021, 125, 9000–9010 CrossRef CAS PubMed.
- S. Lin, Z. Pei, B. Zhang, H. Ma and W. Liang, J. Phys. Chem. A, 2022, 126, 239–248 CrossRef CAS.
- S. Metz and C. M. Marian, ChemPhotoChem, 2022, 6, e202200098 CrossRef CAS.
- A. Takiguchi, N. Inai, S. Kang, M. Hagai, S. Lee, T. Yanai, D. Kim and H. Shinokubo, Chem. Commun., 2022, 58, 5956–5959 RSC.
- P. Rybczyński, M. H. Bousquet, A. Kaczmarek-Kędziera, B. Jędrzejewska, D. Jacquemin and B. Ośmiałowski, Chem. Sci., 2022, 13, 13347–13360 Search PubMed.
- J. Cerezo and F. Santoro, J. Comput. Chem., 2023, 44, 626–643 CrossRef CAS PubMed.
- K. Shizu and H. Kaji, Nat. Commun., 2024, 15, 4723 Search PubMed.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Nat. Rev. Chem., 2019, 3, 589–604 Search PubMed.
- T. Toyao, Z. Maeno, S. Takakusagi, T. Kamachi, I. Takigawa and K.-i. Shimizu, ACS Catal., 2020, 10, 2260–2297 CrossRef CAS.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 22858–22893 Search PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler and F. Glorius, Chem. Soc. Rev., 2020, 49, 6154–6168 Search PubMed.
- K. Jorner, A. Tomberg, C. Bauer, C. Sköld and P.-O. Norrby, Nat. Rev. Chem., 2021, 5, 240–255 CrossRef CAS.
- P. Schwaller, A. C. Vaucher, R. Laplaza, C. Bunne, A. Krause, C. Corminboeuf and T. Laino, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1604 CrossRef.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS.
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343–348 CrossRef CAS.
- S. Singh, M. Pareek, A. Changotra, S. Banerjee, B. Bhaskararao, P. Balamurugan and R. B. Sunoj, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 1339–1345 Search PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. Martinez Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS.
- Z.-J. Zhang, S.-W. Li, J. C. A. Oliveira, Y. Li, X. Chen, S.-Q. Zhang, L.-C. Xu, T. Rogge, X. Hong and L. Ackermann, Nat. Commun., 2023, 14, 3149 CrossRef CAS.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. den Hartog and T. Noël, Science, 2024, 383, eadj1817 CrossRef CAS PubMed.
- A. Yada, K. Nagata, Y. Ando, T. Matsumura, S. Ichinoseki and K. Sato, Chem. Lett., 2018, 47, 284–287 CrossRef CAS.
- J. Werth and M. S. Sigman, J. Am. Chem. Soc., 2020, 142, 16382–16391 CrossRef CAS.
- J. Werth and M. S. Sigman, ACS Catal., 2021, 11, 3916–3922 Search PubMed.
- S. M. Lundberg and S.-I. Lee, in Proceedings of the 31st International Conference on Neural Information Processing Systems, 2017, pp. 4768–4777 Search PubMed.
- S. Mine, Y. Jing, T. Mukaiyama, M. Takao, Z. Maeno, K.-i. Shimizu, I. Takigawa and T. Toyao, Chem. Lett., 2022, 51, 269–273 CrossRef CAS.
- H. Liu, K. Liu, H. Zhu, W. Guo and Y. Li, RSC Adv., 2024, 14, 7276–7282 RSC.
- X. Li, Y. Che, L. Chen, T. Liu, K. Wang, L. Liu, H. Yang, E. O. Pyzer-Knapp and A. I. Cooper, Nat. Chem., 2024, 16, 1286–1294 CrossRef CAS PubMed.
- H. Nakajima, C. Murata, N. Noto and S. Saito, J. Org. Chem., 2025, 90, 1054–1060 CrossRef CAS PubMed.
- T. Nagano, D. B. Bagal, R. Kunisada, H. Nakajima, K. Sano, N. Noto and S. Saito, Chem.–Eur. J., 2025, 31, e202500955 CrossRef CAS PubMed.
- D. Dalmau, M. S. Sigman and J. V. Alegre-Requena, Chem. Sci., 2025, 16, 8555–8560 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem.–Eur. J., 2021, 27, 9934–9947 CrossRef CAS PubMed.
- X. Li, P. M. Maffettone, Y. Che, T. Liu, L. Chen and A. I. Cooper, Chem. Sci., 2021, 12, 10742–10754 RSC.
- L.-C. Yang, X. Li, S.-Q. Zhang and X. Hong, Org. Chem. Front., 2021, 8, 6187–6195 RSC.
- S. K. Kariofillis, S. Jiang, A. M. Żurański, S. S. Gandhi, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 1045–1055 CrossRef CAS.
- N. Noto, A. Yada, T. Yanai and S. Saito, Angew. Chem., Int. Ed., 2023, 62, e202219107 CrossRef CAS.
- L. Schlosser, D. Rana, P. Pflüger, F. Katzenburg and F. Glorius, J. Am. Chem. Soc., 2024, 146, 13266–13275 CrossRef CAS.
- L. Dai, Y. Fu, M. Wei, F. Wang, B. Tian, G. Wang, S. Li and M. Ding, J. Am. Chem. Soc., 2024, 146, 19019–19029 Search PubMed.
- L.-Y. Fan, X.-T. Li, X.-X. Luo, B. Zhu and W. Guan, Chem.–Eur. J., 2025, 31, e202500935 CrossRef CAS.
- N. Noto, R. Kunisada, T. Rohlfs, M. Hayashi, R. Kojima, O. García Mancheño, T. Yanai and S. Saito, Nat. Commun., 2025, 16, 3388 CrossRef CAS.
- N. Inai, S. Yamaguchi and T. Yanai, ACS Phys. Chem. Au, 2023, 3, 540–552 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 Search PubMed.
- R. Ishimatsu, S. Matsunami, K. Shizu, C. Adachi, K. Nakano and T. Imato, J. Phys. Chem. A, 2013, 117, 5607–5612 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 Search PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS.
- H. Sun, C. Zhong and J.-L. Brédas, J. Chem. Theory Comput., 2015, 11, 3851–3858 Search PubMed.
- M. Hagai, N. Inai, T. Yasuda, K. J. Fujimoto and T. Yanai, Sci. Adv., 2024, 10, eadk3219 CrossRef CAS.
- D. Hall, J. C. Sancho-García, A. Pershin, G. Ricci, D. Beljonne, E. Zysman-Colman and Y. Olivier, J. Chem. Theory Comput., 2022, 18, 4903–4918 CrossRef CAS.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330–334 CrossRef CAS PubMed.
- L. Yang, Z. Huang, G. Li, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2018, 57, 1968–1972 CrossRef CAS PubMed.
- Y.-Y. Liu, D. Liang, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2019, 55, 4853–4856 Search PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS.
- M. Kudisch, C.-H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479–19486 CrossRef CAS PubMed.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567–9571 CrossRef CAS.
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Beilstein J. Org. Chem., 2014, 10, 1099–1106 Search PubMed.
- R. Honeker, R. A. Garza-Sanchez, M. N. Hopkinson and F. Glorius, Chem.–Eur. J., 2016, 22, 4395–4399 CrossRef CAS.
- W. Kong, C. Yu, H. An and Q. Song, Org. Lett., 2018, 20, 349–352 CrossRef CAS PubMed.
- X.-H. Ouyang, Y. Li, R.-J. Song and J.-H. Li, Org. Lett., 2018, 20, 6659–6662 Search PubMed.
- N. Noto, Y. Tanaka, T. Koike and M. Akita, ACS Catal., 2018, 8, 9408–9419 CrossRef CAS.
- N. Noto, T. Koike and M. Akita, ACS Catal., 2019, 9, 4382–4387 CrossRef CAS.
- D. Koyama, H. J. A. Dale and A. J. Orr-Ewing, J. Am. Chem. Soc., 2018, 140, 1285–1293 CrossRef CAS.
- N. Noto, K. Takahashi, S. Goryo, A. Takakado, K. Iwata, T. Koike and M. Akita, J. Org. Chem., 2020, 85, 13220–13227 CrossRef CAS.
- Y. Matsui, T. Ikeda, Y. Takahashi, M. Kamata, M. Akagi, Y. Ohya, R. Fujino, H. Namai, E. Ohta, T. Ogaki, T. Miyashi, S. Tero-Kubota, K. Mizuno and H. Ikeda, Asian J. Org. Chem., 2017, 6, 458–468 CrossRef CAS.
- T. Furutani, Y. Matsui, R. Hashimoto, H. Ikeda, T. Ogaki, M. Yamawaki, H. Suzuki and Y. Yoshimi, J. Org. Chem., 2025, 90, 4028–4036 CrossRef CAS.
- Y. Tamaki, K. Kamogawa, R. Inoue, P. Ceroni and O. Ishitani, J. Am. Chem. Soc., 2025, 147, 33010–33022 CrossRef CAS.
- T. P. Cheshire, M. K. Brennaman, P. G. Giokas, D. F. Zigler, A. M. Moran, J. M. Papanikolas, G. J. Meyer, T. J. Meyer and F. A. Houle, J. Phys. Chem. B, 2020, 124, 5971–5985 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |