
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyterrylenes: synthesis and regioregularity effect on p-type charge transport and deep-red light photodetection in OFETs
Chitrak
Ghosh†
 a,
Minji
Chung†
b,
Hayeong
Park
b,
Aniket
Jitendra Talreja
a,
Ullrich
Scherf
c,
Joon Hak
Oh
*b and
Suman Kalyan
Samanta
*a
a,
Minji
Chung†
b,
Hayeong
Park
b,
Aniket
Jitendra Talreja
a,
Ullrich
Scherf
c,
Joon Hak
Oh
*b and
Suman Kalyan
Samanta
*a
aDepartment of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: sksamanta@chem.iitkgp.ac.in
bSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul 08826, Republic of Korea. E-mail: joonhoh@snu.ac.kr
cMacromolecular Chemistry Group (buwmakro) and Wuppertal Center for Smart Materials and Systems (CM@S), D-42119 Wuppertal, Germany
First published on 10th December 2025
Abstract
Polymers of higher rylene dyes remain largely unexplored despite their promising photophysical properties, due to the synthetic challenges and poor solubility, in contrast to their well-studied lower homologs, perylene and naphthalene derivatives. In this study, we report the first synthesis of terrylene-based regioregular and regioirregular homopolymers, namely rr-Polyterrylene and ri-Polyterrylene, respectively. Selective bromination of our previously synthesized 7,8-bis(hexadecyloxy)terrylene (TER-C16) furnished the corresponding dibromoterrylene. Then, Ni(0)-mediated Yamamoto homocoupling reaction produced the first soluble terrylene-based homopolymers. Both polymers achieved high solubility, high thermal stability, alongside very high molecular weight. Owing to these properties, the polymers were successfully deployed in organic field-effect transistors (OFETs), achieving p-type mobility 27 times higher than the monomeric unit TER-C16. Moreover, the photoresponsive OFETs from rr-Polyterrylene demonstrated 116-times improved photodetection ability compared to TER-C16 with specific detectivity (D*) of 4.3 × 1010 Jones in the deep red region, which is the highest among terrylenes and their derivatives. This work introduced the synthesis of dibromoterrylene, a versatile synthetic intermediate with significant potential for constructing diverse polymers. The strategic functionalization of terrylene molecules revealed a novel synthetic approach, enabling innovative polymer construction and facilitating significant advances in organic electronics, specifically OFETs and organic phototransistors (OPTs).
Introduction
Novel polymers synthesized from unique and unexplored monomeric units represent an essential frontier in polymer chemistry and materials science, which are promising for breakthrough applications across multiple domains.1–4 Polymers constructed with lower rylene dyes are well explored in optoelectronic applications such as organic field-effect transistors (OFETs), organic solar cells (OSCs), and organic phototransistors (OPTs) due to their unique molecular structures and distinctive photophysical properties.5 In recent years, conjugated polymers with engineered bandgaps have been extensively investigated for OFETs and OPTs.6 The copolymers comprised of NDI and PDI are well-explored in organic field effect transistors and have achieved high mobility.7–10 Compared to their imide analogues (NDI and PDI), perylene- and naphthalene-based polymers remain substantially less explored despite their considerable potential in device applications.11,12 Beyond the rylene family, numerous conjugated homopolymers have demonstrated high hole mobilities when implemented in OFETs.13 Among the conjugated homopolymers, poly(3-hexyl)thiophenes,14 poly(3-hexyl)selenophene,15 poly(4,8-didodecylbenzo[1,2-b:4,5-b′]dithiophene),16 polyindolo[3,2-b]carbazoles17 have shown efficient p-type mobilities in OFET devices. Notably, the regioregularity of conjugated polymers exhibits a direct correlation with their charge carrier mobility in OFETs. For instance, poly(3-hexylthiophene-2,5-diyl) (P3HT), widely used in OFET devices, has large differences in charge transport properties based on the regioregularity. Regioregular P3HT demonstrated over 104 times mobility compared to regioirregular P3HT due to planarization of the backbone and solid-state self-assembly to form well-defined, organized three-dimensional polycrystalline structures.14–18 Higher rylene analogues, particularly terrylene and its derivatives, remain underexplored compared to their lower analogues due to persistent synthetic challenges and solubility limitations. Research on higher rylene derivatives for optoelectronic applications, particularly OFETs, remains extremely limited, with only a handful of scientific studies exploring their potential.19–21 After Clar's initial synthesis in 1948,22 research on terrylenes primarily focused on developing substituted derivatives, although researchers continued to face significant synthetic challenges.23–26 We recently developed an innovative, low-cost synthetic approach for 7,8-bis(alkyloxy)terrylenes, successfully addressing previous challenges of synthetic complexity and solubility limitations.27The 7,8-bis(alkyloxy)terrylenes demonstrated efficient p-type mobility and high photodetection ability, yet functionalization of the terrylene core remains less explored. Despite extensive synthesis of small TDI molecules across various applications, polymers incorporating terrylene diimide (TDI) remain remarkably underexplored in scientific research.28 To date, TDI-based polymers are rare in the scientific literature, underscoring the substantial potential for innovative research and development in this emerging field of polymer chemistry.29 In this context, apart from TDI, polymers of terrylene core have never been realized before.
Herein, we have synthesized two novel homopolymers of 7,8-bis(hexadecyloxy)terrylene (TER-C16), namely, rr-Polyterrylene and ri-Polyterrylene for the first time (Fig. 1). The synthesis involves bromination of TER-C16 to obtain dibromoterrylenes, marking functionalization of the bay-substituted terrylene core, which hold significant potential as versatile building blocks for synthesizing a diverse range of small molecules and polymers. A facile Ni(0)-mediated Yamamoto homocoupling polymerization of the dibromoterrylenes provided rr-Polyterrylene and ri-Polyterrylene based on regioregularity. The synthesized homopolymers demonstrated a notable bathochromic shift and reduced optical bandgap relative to the parent monomer, making these materials exceptionally promising for advanced optoelectronic devices, OFETs, and OPTs. The OFET devices fabricated from these polymers showed efficient p-type charge transport reaching up to 0.001 cm2 V−1 s−1 for rr-Polyterrylene, which is 27-times higher than its monomer TER-C16. The extended absorption spectra of these polyterrylenes demonstrate great potential for deep red-light detection, which is crucial for various applications in biomedicine or agriculture. In biomedical science, light within the therapeutic window (600–1000 nm) penetrates deeply into tissues with minimal absorption by blood, making it ideal for deep-tissue imaging and light-based therapies that reduce inflammation and promote healing.30–32 Specifically, 670 nm light has been shown to enhance adenosine triphosphate (ATP) production in mitochondria, offering potential benefits for neurodegenerative disease treatment.33 In agriculture, chlorophyll strongly absorbs light around 670 nm, optimizing photosynthesis and promoting plant growth, particularly in a controlled environment.34 Light-driven OFETs were examined with these polymers and achieved a high specific detectivity (D*) of 4.3 × 1010 Jones in the deep red region, which is so far the highest value among terrylene and its derivatives. Regioregular polymer performed better than regioirregular polymer in both OFET and OPT devices. This work demonstrated not only the high potential of terrylene-based polymers in advanced optoelectronic domains but also opened a new direction in higher rylene chemistry.
 | ||
| Fig. 1 Schematic representation of functionalization of terrylene. Vial pictures showing the solutions of compounds 1, 2 and rr-Polyterrylene in chloroform under daylight. | ||
Results and discussion
Synthesis of polyterrylenes
Synthesis of polyterrylenes was accomplished by brominating terrylenes to generate dibrominated intermediates, followed by a Ni(0)-mediated Yamamoto homocoupling reaction, which furnished the conjugated peri-connected terrylene polymers (Scheme 1). At first, TER-C16 was synthesized using our previously reported synthetic method.27 After that, a facile bromination was performed with liquid bromine to synthesize dibromo derivatives of TER-C16 with 95% yield. Notably, more than one dibromo derivative was obtained in this process, which was confirmed by 1H NMR analysis (Fig. S1), indicating the formation of regioisomers. A similar synthetic outcome was observed when employing N-bromosuccinimide (NBS) as a brominating agent instead of liquid bromine. Moreover, using two equivalents of the brominating agent, MALDI-ToF mass spectrometric analyses revealed the absence of either monobrominated or tribrominated terrylene derivatives (Fig. S2a). Following extensive column chromatographic purification, only compound 2 was successfully isolated, characterized by its relatively higher polarity. The other isomer (3) consistently co-eluted with compound 2, rendering its complete separation highly challenging despite multiple repetitive column chromatographic attempts. Comprehensive structural characterization of compound 2 was achieved through MALDI-ToF mass spectrometry (Fig. S2) and NMR spectroscopy (Fig. S3–S5). Analysing Fig. S1 and S3 carefully, the ratio of pure compound 2 to 3 was found to be nearly 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. All attempts to grow a diffraction-quality single crystal of compound 2 remained unsuccessful. In the final synthesis step, a homocoupling reaction of compound 2 under Yamamoto conditions using Ni(COD)2 facilitated the formation of terrylene homopolymer, designated as rr-Polyterrylene. Surprisingly, the polymerization proceeds with exceptional rapidity, achieving substantial polymer growth within just 15 minutes, resulting in a polymer that becomes predominantly insoluble in chloroform and chlorobenzene, even at higher temperatures, despite the presence of two long-chain hexadecyloxy (–OC16H33) substituents. In this straightforward method, the polymerization yielded a limited soluble fraction (<20%) with exceptionally high weight average molecular weight (Mw) (Table S1).
:1. All attempts to grow a diffraction-quality single crystal of compound 2 remained unsuccessful. In the final synthesis step, a homocoupling reaction of compound 2 under Yamamoto conditions using Ni(COD)2 facilitated the formation of terrylene homopolymer, designated as rr-Polyterrylene. Surprisingly, the polymerization proceeds with exceptional rapidity, achieving substantial polymer growth within just 15 minutes, resulting in a polymer that becomes predominantly insoluble in chloroform and chlorobenzene, even at higher temperatures, despite the presence of two long-chain hexadecyloxy (–OC16H33) substituents. In this straightforward method, the polymerization yielded a limited soluble fraction (<20%) with exceptionally high weight average molecular weight (Mw) (Table S1).
 | ||
| Scheme 1 Synthesis of terrylene-based homopolymers, rr-Polyterrylene and ri-Polyterrylene. | ||
To address the solubility problem, 10 mol% of 1-bromonaphthalene was introduced alongside dibromoterrylenes to truncate the extensive polymer growth and obtain well-soluble polyterrylenes as major fractions. In this modified method, the polymerization reactions were continued for 30 min in the presence of 10 mol% 1-bromonapthalene, and then, 20 mol% of 1-bromonaphthalene was added again as the end-cap, and the reaction was continued for an additional 2 h. This synthetic approach successfully generated highly soluble polyterrylenes as deep blue solids with high isolated yields. The pure dibromoterrylene (2) afforded a regioregular polymer, rr-Polyterrylene, with a 77% yield. However, the unseparated mixture of compounds 2 and 3, produced the regioirregular polymer, ri-Polyterrylene, with a 71% yield. Both polymers showed high solubility across a range of common organic solvents, including chloroform, toluene, chlorobenzene and o-dichlorobenzene. Both of the synthesized polymers were characterized by 1H nuclear magnetic resonance (NMR) (Fig. S6 and S7) along with gel permeation chromatography (GPC), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC) studies. The GPC analyses determined the weight average molecular weight (Mw) of 177 kg mol−1 for rr-Polyterrylene and 122 kg mol−1 for ri-Polyterrylene (Table 1). This result demonstrated the utility of this strategy for synthesizing soluble terrylene homopolymers with high molecular weight. TGA analyses revealed exceptional thermal stability for both polymers, with the onset of 5% weight reduction under nitrogen occurring at 316 °C and 304 °C for rr-Polyterrylene and ri-Polyterrylene, respectively (Fig. S8). Such high thermal stability enables polymers to perform consistently across various optoelectronic devices. The DSC analysis revealed no distinct peak due to the low crystallinity and high molecular weight of the polymers (Fig. S9).
| Compounds | λ max,sol (nm) | ε (L mol−1 cm−1) | λ max,film (nm) | E g,opt (eV) | EHOMOa (eV) | E LUMO (eV) | M n (kg mol−1) | M w (kg mol−1) | PDI | T d (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Obtained from cyclic voltammogram. | ||||||||||
| TER-C16 | 544 | 65900 |
587 | 1.96 | −5.19 | −3.24 | — | — | — | 322 |
| rr-Polyterrylene | 607 | 66000 |
609 | 1.85 | −5.28 | −3.43 | 66.13 | 177.15 | 2.6 | 316 |
| ri-Polyterrylene | 604 | 64000 |
607 | 1.87 | −5.30 | −3.43 | 46.61 | 122.32 | 2.6 | 304 |
Photophysical and redox properties
To elucidate the fundamental optoelectronic characteristics of the novel polyterrylenes, we performed comprehensive spectroscopic characterization via UV-vis absorption spectroscopy in both solutions (10 µM in chloroform) and thin films (Fig. 2a). Notably, TER-C16 demonstrated an absorption maximum (λmax) at 544 nm, indicating a 0–0 transition, which is a characteristic feature of terrylene.27 The pure dibromoterrylene (2) showed λmax at 558 nm, with a slight red-shift relative to TER-C16. Interestingly, rr-Polyterrylene and ri-Polyterrylene exhibited prominent bathochromic shifts, presenting λmax at 607 nm and 604 nm, respectively. The bathochromic shift of over 60 nm reveals pronounced π-electron delocalization and expanded conjugation, offering compelling evidence of extensive structural and electronic transformations resulting from polymerization. In thin films, the absorption spectra showed minimal red-shift (Table 1) and notable changes at the trail as compared to their solution, indicating a possible aggregation in the solid state (Fig. 2a). From the onset of the absorbance spectra, the optical band gap of the polymers was found to be 1.85 eV and 1.87 eV for rr-Polyterrylene and ri-Polyterrylene, respectively. | ||
| Fig. 2 Photophysical and redox properties of polyterrylenes. (a) Normalized UV-vis spectra in 10 µM chloroform solutions and in thin films (inset: image showing the spectral changes of rr-Polyterrylenes and ri-Polyterrylenes in both solutions and in thin films) and (b) cyclic voltammograms of rr-Polyterrylene and ri-Polyterrylene in dry DCM solution at a sweeping rate of 0.05 V s−1 using ferrocene as a standard. | ||
To investigate the redox properties, cyclic voltammetry experiments were performed in dichloromethane solutions of the polymers (Fig. 2b). From the oxidation peak, the highest occupied molecular orbitals (HOMO) energy level was calculated as −5.28 eV and −5.30 eV for rr-Polyterrylene and ri-Polyterrylene, respectively.35,36 These near-identical HOMO energy levels suggest minimal electronic structure variations between the two polyterrylenes, indicating that the regioisomeric configuration exerts a negligible impact on the frontier molecular orbital energetics. The lowest unoccupied molecular orbital (LUMO) energy levels were calculated from the highest occupied molecular orbital (HOMO) energy and the optical band gap. The resulting LUMO energy levels at −3.43 eV for both rr-Polyterrylene and ri-Polyterrylene demonstrated minimal variation between polymer configurations. However, the LUMO energy levels of the polymers are significantly low-lying compared to those of TER-C16.27
Theoretical studies
Theoretical investigations were performed using the density functional theory (DFT)/B3LYP method with a 6-31G basis set in Gaussian 16 (Fig. 3, Table S2). A simplified model compound featuring the terrylene core functionalized with methoxy (–OCH3) substituents was selected for computational analysis, with three terrylene moieties connected through the peri-position and was considered as the model polymeric unit to enable detailed computational investigation of the molecular structure. Geometry optimization revealed significant deviation from molecular planarity among terrylene moieties, indicating structural perturbation induced by peri-substitution. The dihedral angle between two terrylene subunits in the peri-position is about 70.2° as the other two remaining hydrogen atoms at the peri-position of terrylene prevent polyterrylene from achieving a fully planar polymer configuration (Fig. 3a). This deviation could help to achieve their high solubility in chlorinated solvents. Theoretical calculations demonstrated that the HOMO and LUMO electron densities are localized along the peripheral π-conjugated backbone of the terrylene framework, displaying antisymmetric distribution (Fig. 3b).37 In context with DFT calculations, polyterrylene showed a slightly uplifted HOMO and low-lying LUMO energy levels compared to 7,8-bis(methyloxy)terrylene, delineating a modest reduction of the band gap of 0.12 eV, which correlates well with their experimental bandgap differences.27 The charge distribution across the bay-functionalized polyterrylene revealed an electronically neutral character predominantly localized within the aromatic core (Fig. 3c), similar to its monomer.27 The HOMO−1 and LUMO+1 energies are also calculated and it is observed that these energies are close to the energies for HOMO and LUMO, respectively (Fig. S10). | ||
| Fig. 3 DFT calculations using the Gaussian16 program with the 6-31G basis set using the B3LYP method showing (a) optimized structure, (b) HOMO and LUMO distribution, and (c) charge distribution of rr-Polyterrylene. | ||
Thin film microstructural analysis
The morphological and microstructural features of the rr-Polyterrylene and ri-Polyterrylene were analyzed using non-contact mode atomic force microscopy (AFM) (Fig. 4a and b) and grazing incidence X-ray diffraction (GIXD) analyses (Fig. 4c and d). Thin films of the rr-Polyterrylene and rr-Polyterrylene were prepared by spin coating a 5 mg mL−1 chloroform solution onto n-octadecyltrimethoxysilane (OTS)-modified SiO2/Si wafer, followed by annealing at 180 °C and 150 °C, respectively, under a nitrogen atmosphere. In contrast to the monomeric unit TER-C16, which exhibited a rod-like fiber morphology, both polymer films displayed a nodular structure38 with significantly reduced crystallinity. This reduction in crystallinity can be attributed to the extended polymer chains, which disrupt long-range molecular ordering. Despite the decrease in overall crystallinity, UV-vis absorption spectroscopy revealed a significant red shift in these polymers compared to the monomer (Fig. 2a) and these spectral changes indicate a structural transformation due to enhanced π–π conjugation.39–41 | ||
| Fig. 4 Device performances and morphological properties. AFM images of (a) rr-Polyterrylene and (b) ri-Polyterrylene, respectively (scale bar = 500 µm). GIWAXS images of (c) rr-Polyterrylene and (d) ri-Polyterrylene, respectively. Transfer curves of (e) rr-Polyterrylene and (f) ri-Polyterrylene, respectively. Output curves of (g) rr-Polyterrylene and (h) ri-Polyterrylene, respectively. | ||
To further investigate how regioregularity affects molecular packing, GIXD analysis was conducted (Fig. 4c and d), and the corresponding parameters are summarized in Table 2. Both films exhibit (010) diffraction peaks at ∼15.22 nm−1, indicating an identical π–π stacking distance of ∼0.41 nm due to their shared backbone structure. The coherence length (Lc) calculated from the full width at half maximum (FWHM) of the (010) reflections reveal that rr-Polyterrylene possesses a larger Lc value (4.83 nm) compared to ri-Polyterrylene (4.25 nm). The increased Lc indicates that higher regioregularity promotes more efficient interchain packing, which enhances exciton migration and charge carrier transport.42,43 Although the crystallinity of the polymer thin films was reduced, partially ordered domains were more densely interconnected, facilitating effective charge transport.44–46
| Compounds | q 010 (nm−1) | d-spacing (nm) | FWHM (nm−1) | Coherence length (nm) |
|---|---|---|---|---|
| ri-Polyterrylene | 15.22 | 0.41 | 1.34 | 4.25 |
| rr-Polyterrylene | 15.22 | 0.41 | 1.18 | 4.83 |
OFET device application
To evaluate the charge-transport properties of the rr-Polyterrylene and ri-Polyterrylene, bottom-gate top-contact field-effect transistors (FETs) were fabricated on OTS-modified SiO2/Si substrates. These devices were characterized in a nitrogen-filled glovebox after being annealed at various temperatures (from 150 °C to 210 °C) for 10 min. Both polymers showed p-type dominant behavior in the N2 atmosphere. The hole mobilities at different annealing temperatures are summarized in Table S3, presenting a comparison of the hole mobilities (µh,avg), on/off ratios (Ion/Ioff), and threshold voltages (Vth). The transfer and output characteristics at the optimized annealing temperatures of 180 °C for rr-Polyterrylene and 150 °C for ri-Polyterrylene are shown in Fig. 4c–f. Compared to TER-C16, which exhibited a relatively low hole mobility of 3.6 × 10−5 cm2 V−1 s−1, the rr-Polyterrylene and ri-Polyterrylene demonstrated significantly enhanced hole mobilities of 1.0 × 10−3 and 2.6 × 10−4 cm2 V−1 s−1, respectively. This considerable improvement in charge carrier mobility indicates more efficient charge transport facilitated by extended π-conjugation and lowered bandgap demonstrated by the polymers compared to the monomer.47,48 Notably, the hole mobility of rr-Polyterrylene was nearly four times greater than that of ri-Polyterrylene, underscoring the critical role of regioregularity in determining the crystallinity and electrical properties of the polymer.49–53 The regioirregular polymer, characterized by its lower molecular weight and marginally increased bandgap, exhibited disrupted intermolecular organization that resulted in substantially reduced hole mobility.54Phototransistor applications
The optoelectronic performance of rr-Polyterrylene and ri-Polyterrylene was further investigated in organic phototransistor (OPT) devices under monochromatic light illumination at 635 nm with an intensity of 18.8 mW cm−2 in vacuum conditions. Fig. 5a and b illustrates the transfer characteristics of the rr-Polyterrylene and ri-Polyterrylene under both dark and light illumination at an applied drain voltage of −80 V. Both polymers exhibited an increase in drain current and a positive threshold voltage shift, indicating easier turn-on under p-channel operation. This behavior is attributed to a photodoping effect, wherein photoexcited charge carriers reduce trap sites.55,56 To quantify the photosensitivity of OPTs, we calculated the photo-current/dark-current ratio (P) and photoresponsivity (R) based on the transfer curves.
where Idark and Ilight represent the drain current measured in dark and under illumination, respectively, Iph is the photocurrent, and Pinc is the incident illumination power on the device channel. Additionally, the external quantum efficiency (EQE, η) of OPTs was calculated, which quantifies the ratio of photogenerated carriers that effectively enhance the drain current to the number of photons incident on the OPT channel using the equation,

Where h is Planck's constant, c is the speed of light, e is the fundamental unit of charge, A is the area of the transistor channel, and λ peak is the peak wavelength of the incident light. The detectivity (D*) was also evaluated, indicating the smallest detectable signal and enabling comparisons across different phototransistor devices. The detectivity was calculated using the following equation:


where A is the phototransistor active area, NEP is the noise equivalent power, and

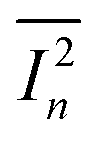 is the measured noise current. When shot noise from the dark drain current is the dominant limitation, D* simplifies to:
is the measured noise current. When shot noise from the dark drain current is the dominant limitation, D* simplifies to:
 | ||
| Fig. 5 Photoresponse characteristics under 635 nm light illumination conditions. Transfer characteristics of (a) rr-Polyterrylene and (b) ri-Polyterrylene under dark and illumination conditions. Vds = −80 V. Calculated (c) photosensitivity, responsivity, and (d) external quantum efficiency, detectivity as a function of the gate voltage. | ||
Fig. 5c and d illustrates the variations in P, R, EQE, and D* as functions of Vgs under light irradiation, with the maximum values summarized in Table S4. Compared to TER-C16, which exhibited a maximum P of 1.6 × 102, R of 7.2 × 10−4 A W−1, EQE of 0.17%, and D* of 3.7 × 108 Jones at 532 nm (79.2 mW cm−2), the polymer thin films demonstrated superior optoelectronic performance. In particular, rr-Polyterrylene exhibited significantly higher values, with a maximum P of 1.6 × 104, R of 5.8 × 10−3 A W−1, EQE of 1.1%, and D* of 2.0 × 1010 Jones at 635 nm (25.5 mW cm−2). This difference between the monomer and regioregularly polymerized materials arises from a structural transition from monomer to polymer, which enhances π-conjugation and molecular ordering, thereby reducing exciton binding energy and promoting efficient exciton dissociation. In contrast, ri-Polyterrylene exhibited lower optoelectronic performance than rr-Polyterrylene, with an R of 6.2 × 10−4 A W−1 and an EQE of 0.12%, values even lower than those of TER-C16. These inferior properties are likely due to a higher density of relatively deep trap states,57–60 which hinder charge transport, coupled with the lower light intensity of the 635 nm laser.61,62
To elucidate the correlation between trap states and optoelectronic performance in the regioisomeric polymers, response and decay times (τr and τd) were analyzed (Fig. 6). The photodetector response time is defined as the duration required to reach 90% of the maximum photocurrent during a rapid change, while the decay time refers to the time needed for the photocurrent to decrease to 10% of its maximum value.63 The decay time is directly related to the density of deep traps due to the photogating effect,64 where trapped charges modulate channel conductance even after the light is turned off, leading to persistent photocurrent and prolonged decay. Additionally, a long rise time indicates that photogenerated electron–hole pairs become trapped before reaching the electrodes, resulting in a slow accumulation of free carriers.65
 | ||
| Fig. 6 Photoresponse characteristics under 635 nm light illumination conditions. Time-dependent photoresponse characteristics of (a) rr-Polyterrylene and (b) ri-Polyterrylene under periodic dark and light illumination conditions, respectively. Recorded photocurrent over time for (c) dark-to-illumination and (d) illumination-to-dark conditions. | ||
Under illumination, rr-Polyterrylene exhibited a significantly shorter response with the corresponding decay times of 0.45 s and 0.39 s, respectively, compared to ri-Polyterrylene, which exhibited values of 0.79 s and 2.54 s. The prolonged decay time in ri-Polyterrylene suggests an increased density of localized trap states that capture photogenerated carriers, leading to slower carrier transport and reduced EQE.66 These trap states act as recombination centers, lowering the number of available photogenerated carriers. In contrast, rr-Polyterrylene demonstrated significantly improved photodetection performance, characterized by a fast response time, highlighting its potential as an organic photodetector material. rr-Polyterrylene features reduced charge localization and minimized trap-induced recombination delays67 which results in weaker polaron localization and enhanced charge transport. Furthermore, the excitons in rr-Polyterrylene diffuse more efficiently through extended crystalline domains, enabling more effective energy transfer and superior photoresponse.68,69
Enhanced π–π stacking and improved molecular ordering further contribute to greater electronic delocalization and weaker electron–phonon coupling, thereby enhancing its optoelectronic properties. Compared to TER-C16, polymerized rr-Polyterrylene and ri-Polyterrylene show potential as active materials for deep-red light sensors due to their bathochromic shift in the absorption spectrum, which extends the absorption range toward lower-energy light.70 To evaluate the deep-red light photodetection performance of these terrylene-based polymers, we compared the drain current under dark and illuminated conditions and measured the variations in P, R, EQE, and D* as functions of Vgs under 670 nm light irradiation (Fig. 7, Table 3). In particular, rr-Polyterrylene exhibited outstanding optoelectronic performance, achieving a maximum P of 7.4 × 103, R of 1.2 × 10−2 A W−1, EQE of 2.2%, and D* of 4.3 × 1010 Jones under monochromatic 670 nm illumination (1.15 mW cm−2) in vacuum conditions. The superior sensing properties of this regioregular polymer are attributed to its low energy disorder and improved overlap between polymer chains. These findings demonstrate that rr-Polyterrylene exhibits high efficiency in deep-red light detection.
 | ||
| Fig. 7 Photoresponse characteristics of rr-Polyterrylene under 670 nm light illumination conditions. (a) Transfer characteristics under dark and 670 nm light illumination conditions. Vds = −80 V. (b) Time-dependent photoresponse characteristics under periodic dark and light illumination conditions. Calculated (c) photosensitivity, responsivity and (d) external quantum efficiency, detectivity as a function of the gate voltage. | ||
| Compounds | P MAX | R MAX (A W−1) | EQEMAX (%) |
|
|---|---|---|---|---|
| rr-Polyterrylene | 7.4 × 103 | 1.2 × 10−2 | 2.200 | 4.3 × 1010 |
| ri-Polyterrylene | 1.8 × 102 | 1.9 × 10−3 | 0.036 | 7.0 × 108 |

Conclusions
In summary, we report the funtionalization of terrylene as a pure dibromoterrylene derivative (compound 2), which demonstrated a pivotal synthetic intermediate for constructing polymers. We also synthesized two terrylene-based homopolymers, rr-Polyterrylene and ri-Polyterrylene, based on regioregularity, for the first time. In polymers, the pronounced π-conjugation resulted in a significant bathochromic shift in the absorption spectrum compared to the parent terrylene monomer. Despite minimal variations in optoelectronic properties arising from regioregularity, substantial differences in device performance were observed when these polymers were implemented in OFET and OPT devices. The regioregular polymer, rr-Polyterrylene, exhibited superior charge transport properties, demonstrating enhanced hole mobility and more pronounced photoresponsive characteristics compared to the ri-Polyterrylene. rr-Polyterrylene also demonstrated the p-type mobility 27-fold higher than its monomer TER-C16 and achieved nearly 116-times higher specific detectivity (D*) of 4.3 × 1010 Jones in photoresponsive OFET devices, which significantly surpasses the performance of previously reported terrylene small molecules. In particular, our regioregular polymer exhibited exceptional deep-red light detection performance, making it highly suitable for various applications. This work opened a new beginning in higher rylene chemistry as it introduced a novel dibromoterrylene synthetic intermediate with exceptional versatility, enabling strategic molecular architecture modifications that will advance organic electronics through innovative polymer construction and developments in OFETs and OPTs.Author contributions
C. G. executed the synthesis, characterization, and studies. M. C. and H. P. performed the thin film characterization, device fabrication, and testing. A. J. T. synthesized some of the starting materials. J. H. O. supervised the thin-film characterization and device experiments. S. K. S. conceptualized the project, designed the synthetic route of polyterrylenes, and supervised all experiments. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc06452j.Acknowledgements
S. K. S. acknowledges SERB, DST (SRG/2019/000922) for funding this research work. S. K. S. Acknowledges the Alexander von Humboldt Foundation, Germany. C. G. is thankful to the Ministry of Education (MoE) for fellowships. We thank the Central Research Facility (CRF), Indian Institute of Technology Kharagpur, for the instrumental facilities. We thank Paramshakti Supercomputing Facility, IIT Kharagpur, for computational studies, and Central Research Facility (CRF), IIT Kharagpur, for the instrumental facilities. This work was supported by the National Research Foundation of Korea (NRF) grant (2023R1A2C3007715, RS-2024-00398065) through the NRF by the Ministry of Science and ICT (MSIT), Korea. This work was also supported by the Technology development Program (RS-2025-25460624) funded by the Ministry of SMEs and Startups (MSS), Korea. The Institute of Engineering Research at Seoul National University provided research facilities for this work.References
- A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS.
- K. Maity, S. Sau, F. Banerjee and S. K. Samanta, ACS Appl. Mater. Interfaces, 2024, 16, 50834–50845 CrossRef CAS PubMed.
- K. Maity, S. Sau and S. K. Samanta, Chem.–Asian J., 2025, 20, e202401053 CrossRef CAS PubMed.
- F. Banerjee and S. K. Samanta, Mater. Chem. Front., 2023, 7, 689–697 RSC.
- P. Cheng, X. Zhao and X. Zhan, Acc. Mater. Res., 2022, 3, 309–318 CrossRef CAS.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- S. K. Samanta, I. Song, J. H. Yoo and J. H. Oh, ACS Appl. Mater. Interfaces, 2018, 10, 32444–32453 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng, H. Yan and A. Facchetti, J. Am. Chem. Soc., 2009, 131, 8–9 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- M. J. Sung, A. Luzio, W.-T. Park, R. Kim, E. Gann, F. Maddalena, G. Pace, Y. Xu, D. Natali, C. de Falco, L. Dang, C. R. McNeill, M. Caironi, Y.-Y. Noh and Y.-H. Kim, Adv. Funct. Mater., 2016, 26, 4984–4997 CrossRef CAS.
- S. K. Samanta, G. S. Kumar, U. K. Ghorai, U. Scherf, S. Acharya and S. Bhattacharya, Macromolecules, 2018, 51, 8324–8329 CrossRef CAS.
- J.-H. Kim, H. U. Kim, D. Mi, S.-H. Jin, W. S. Shin, S. C. Yoon, I.-N. Kang and D.-H. Hwang, Macromolecules, 2012, 45, 2367–2376 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- M. Heeney, W. Zhang, D. J. Crouch, M. L. Chabinyc, S. Gordeyev, R. Hamilton, S. J. Higgins, I. McCulloch, P. J. Skabara, D. Sparrowe and S. Tierney, Chem. Commun., 2007,(47), 5061–5063 RSC.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, Chem. Mater., 2006, 18, 3237–3241 CrossRef CAS.
- Y. Li, Y. Wu and B. S. Ong, Macromolecules, 2006, 39, 6521–6527 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- C. Liu, Z. Liu, H. T. Lemke, H. N. Tsao, R. C. G. Naber, Y. Li, K. Banger, K. Müllen, M. M. Nielsen and H. Sirringhaus, Chem. Mater., 2010, 22, 2120–2124 CrossRef CAS.
- G. Battagliarin, S. R. Puniredd, S. Stappert, W. Zajaczkowski, S. Wang, C. Li, W. Pisula and K. Müllen, Adv. Funct. Mater., 2014, 24, 7530–7537 CrossRef CAS.
- K. Zhao, Y.-C. Xu, J.-C. Zeng, Y.-K. Qu, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2025, 64, e202503571 CrossRef CAS PubMed.
- E. Clar, Chem. Ber., 1948, 81, 52–63 CrossRef CAS.
- A. Bohnen, K.-H. Koch, W. Lüttke and K. Müllen, Angew Chem. Int. Ed. Engl., 1990, 29, 525–527 CrossRef.
- U. Anton, M. Adam, M. Wagner, Z. Qi-Lin and K. Müllen, Chem. Ber., 1993, 126, 517–521 CrossRef CAS.
- Y. Avlasevich, C. Kohl and K. Müllen, J. Mater. Chem., 2006, 16, 1053–1057 RSC.
- A. Hirono, H. Sakai and T. Hasobe, Chem.–Asian J., 2019, 14, 1754–1762 CrossRef CAS PubMed.
- C. Ghosh, H. Park, A. Hazra, B. Mondal, M. Chung, U. Scherf, J. H. Oh and S. K. Samanta, J. Mater. Chem. C, 2025, 13, 7518–7527 RSC.
- L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- T. L. Andrew and T. M. Swager, Macromolecules, 2011, 44, 2276–2281 CrossRef CAS PubMed.
- Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC.
- G. E. Glass, Aesthet. Surg. J., 2022, 42, 566 CrossRef PubMed.
- S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC.
- P. Kaynezhad, I. Tachtsidis and G. Jeffery, Exp. Eye Res., 2016, 152, 88–93 CrossRef CAS PubMed.
- S. A. D. M. Zahir, A. F. Omar, M. F. Jamlos, M. A. M. Azmi and J. Muncan, Sens. Actuators, A, 2022, 338, 113468 CrossRef CAS.
- B. Mondal, R. Tiwari, S. Manna, F. Banerjee, R. Singh and S. K. Samanta, ACS Appl. Energy Mater., 2025, 8, 3459–3469 CrossRef CAS.
- S. Manna, P. Kumar, R. Tiwari, B. Mondal, C. Ghosh, R. Singh and S. K. Samanta, Energy Fuels, 2025, 39, 10021–10030 CAS.
- V. Sharma, H. Khan, M. Walker, H. Ahmad, A. Thanai, T. Marszalek, D. Schollmeyer, M. Baumgarten, E. W. Evans and A. Keerthi, Chem.–Eur. J., 2024, 30, e202401462 CrossRef CAS PubMed.
- H. N. Tsao and K. Müllen, Chem. Soc. Rev., 2010, 39, 2372–2386 RSC.
- N. J. Hestand and F. C. Spano, J. Chem. Phys., 2015, 143 Search PubMed.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- Y. Yuan, J. Shu, P. Liu, Y. Zhang, Y. Duan and J. Zhang, J. Phys. Chem. B, 2015, 119, 8446–8456 CrossRef CAS PubMed.
- E. Gutiérrez-Meza, R. Malatesta, H. Li, I. Bargigia, A. R. Srimath Kandada, D. A. Valverde-Chávez, S.-M. Kim, H. Li, N. Stingelin, S. Tretiak, E. R. Bittner and C. Silva-Acuña, Sci. Adv., 2021, 7, eabi5197 CrossRef PubMed.
- T. Eder, T. Stangl, M. Gmelch, K. Remmerssen, D. Laux, S. Höger, J. M. Lupton and J. Vogelsang, Nat. Commun., 2017, 8, 1641 CrossRef PubMed.
- K. Janus, D. Chlebosz, A. Janke, W. Goldeman and A. Kiersnowski, Macromolecules, 2023, 56, 964–973 CrossRef CAS.
- J. Mun, Y. Ochiai, W. Wang, Y. Zheng, Y.-Q. Zheng, H.-C. Wu, N. Matsuhisa, T. Higashihara, J. B. H. Tok, Y. Yun and Z. Bao, Nat. Commun., 2021, 12, 3572 CrossRef CAS PubMed.
- Y. Li, W. K. Tatum, J. W. Onorato, Y. Zhang and C. K. Luscombe, Macromolecules, 2018, 51, 6352–6358 CrossRef CAS.
- Z. Applied Physics LettersGan, L. Wang, J. Cai, C. Guo, C. Chen, D. Li, Y. Fu, B. Zhou, Y. Sun, C. Liu, J. Zhou, D. Liu, W. Li and T. Wang, Nat. Commun., 2023, 14, 6297 CrossRef PubMed.
- Y. Liu, H. Wang, S. Li, C. Chen, L. Xu, P. Huang, F. Liu, Y. Su, M. Qi, C. Yu and Y. Zhou, Nat. Commun., 2020, 11, 1724 CrossRef CAS PubMed.
- Y. Kim, H. Park, J. S. Park, J.-W. Lee, F. S. Kim, H. J. Kim and B. J. Kim, J. Mater. Chem. A, 2022, 10, 2672–2696 RSC.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS.
- Y. M. Gross, D. Trefz, R. Tkachov, V. Untilova, M. Brinkmann, G. L. Schulz and S. Ludwigs, Macromolecules, 2017, 50, 5353–5366 CrossRef CAS.
- D. P. McMahon, D. L. Cheung, L. Goris, J. Dacuña, A. Salleo and A. Troisi, J. Phys. Chem. C, 2011, 115, 19386–19393 CrossRef CAS.
- R. Steyrleuthner, R. Di Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245–4256 CrossRef CAS PubMed.
- J.-S. Kim, J.-H. Kim, W. Lee, H. Yu, H. J. Kim, I. Song, M. Shin, J. H. Oh, U. Jeong, T.-S. Kim and B. J. Kim, Macromolecules, 2015, 48, 4339–4346 CrossRef CAS.
- H. Yu, Z. Bao and J. H. Oh, Adv. Funct. Mater., 2013, 23, 629–639 CrossRef CAS.
- L. Cao, C. Ren and T. Wu, J. Mater. Chem. C, 2023, 11, 3428–3447 RSC.
- X. Wang, F. Zhao, Z. Xue, Y. Yuan, M. Huang, G. Zhang, Y. Ding and L. Qiu, Adv. Electron. Mater., 2019, 5, 1900174 CrossRef.
- J. Li, A. Tamayo, A. Quintana, S. Riera-Galindo, R. Pfattner, Y. Gong and M. Mas-Torrent, J. Mater. Chem. C, 2023, 11, 8178–8185 RSC.
- J. A. Carr, M. Elshobaki and S. Chaudhary, Appl. Phys. Lett., 2015, 107, 203302 CrossRef.
- M. Shou, J. Zheng, X. Liu, J. Zhou, Z. Xie, Q. Liao, H. Li and L. Liu, J. Mater. Chem. C, 2025, 13, 2969–2977 RSC.
- C. Labanti, J. Wu, J. Shin, S. Limbu, S. Yun, F. Fang, S. Y. Park, C.-J. Heo, Y. Lim and T. Choi, Nat. Commun., 2022, 13, 3745 CrossRef CAS PubMed.
- Z. Bahrami, K. Schnittker, W. Adi, A. Beisenova, F. Yesilkoy, D. Thompson and J. Andrews, Adv. Opt. Mater., 2024, 12, 2401269 CrossRef CAS.
- I. Song, J. Ahn, S. H. Lee, S. Lee, S. Kim, Y. I. Cho and J. H. Oh, Chem. Eng. J., 2025, 505, 158991 CrossRef CAS.
- J. Shin and H. Yoo, Nanomaterials, 2023, 13, 882 CrossRef CAS PubMed.
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC.
- J. S. Liu, C. X. Shan, B. H. Li, Z. Z. Zhang, C. L. Yang, D. Z. Shen and X. W. Fan, Appl. Phys. Lett., 2010, 97, 251102 CrossRef.
- J. H. Bombile, M. J. Janik and S. T. Milner, Phys. Chem. Chem. Phys., 2018, 20, 317–331 RSC.
- O. V. Mikhnenko, P. W. M. Blom and T.-Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- Y. Zhang, M. T. Sajjad, O. Blaszczyk, A. J. Parnell, A. Ruseckas, L. A. Serrano, G. Cooke and I. D. W. Samuel, Chem. Mater., 2019, 31, 6548–6557 CrossRef CAS.
- H. Watanabe, K. Tanaka and Y. Chujo, Polymers, 2021, 13, 4021 CrossRef CAS PubMed.
Footnote |
| † CG and MC contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |