
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInherently chiral homo-heteracalixarenes: design and synthesis via an enantioselective intramolecular Sonogashira cross-coupling reaction
Y.-F.
Jiang
a,
Y.
Zhang
a,
S.
Tong
 *a,
J.
Zhu
b and
M.-X.
Wang
a
*a,
J.
Zhu
b and
M.-X.
Wang
a
aKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: tongshuo@mail.tsinghua.edu.cn; Web: http://mascl.group
bLaboratory of Synthesis and Natural Products (LSPN), Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédéral de Lausanne, EPFL-SB-ISIC-LSPN, BCH5304, CH-1015 Lausanne, Switzerland
First published on 20th November 2025
Abstract
Recent research has seen growing interest in synthesizing inherently chiral macrocycles, driven by their potential applications in chiral supramolecular chemistry. We present herein our design and synthesis of a series of novel inherently chiral macrocycles. These compounds, termed homo-heteracalixarenes, feature a 1,2-diphenylethyne fragment that replaces one of the aryl-heteroatom-aryl linkages found in classic heteracalix[4]aromatics. De novo macrocyclization of linear achiral substrates via an intramolecular Sonogashira cross-coupling reaction affords the 17- or 18-membered chiral cyclophanes in modest yields with up to 90% ee. The strained bent alkyne moiety within the macrocycle provides a key reactive handle for macrocycle-to-macrocycle derivatization. Further, we showed the unique macrocyclic structures and demonstrated interesting chiroptical properties of the obtained enantioenriched homo-calixarenes and their derivatives.
Introduction
“Inherent chirality” was first introduced by Böhmer in 1994 to characterize chiral calixarenes lacking both a plane of symmetry and an inversion center.1 As later elaborated by Mandolini and Schiaffino in 2004, the introduction of curvature into an idealized planar molecular structure that already lacks symmetry in its bidimensional representation generates inherent chirality.2 This concept has gained widespread acceptance and is now routinely employed in macrocyclic and supramolecular chemistry.3,4 Inherently chiral macrocycles (ICMs) exhibit several distinctive features: (1) they possess a chiral cavity as a defining structural characteristic; (2) their structural diversity is significantly enhanced since the building blocks are not restricted to chiral molecules; (3) the unrestricted selection of constituent units enables more flexible design and precise modulation of chiral cavities with tunable geometries, dimensions, and electronic properties. These distinctive advantages make ICMs promising candidates for applications in enantioselective recognition, asymmetric catalysis, and chiroptical devices. However, the reliance on chiral HPLC separation for obtaining enantiopure ICMs in early studies posed a bottleneck for advancing chiral supramolecular development. Pleasingly, recent advances have revolutionized the enantioselective synthesis of ICMs. Since 2020, our group and others have established catalytic asymmetric strategies, including (dynamic) kinetic resolution5–7 and desymmetrization of prochiral macrocycles.8 These methodologies have enabled the efficient construction of highly enantioenriched inherently chiral macroarchitectures, including calixarenes,8,9 resorcinarenes,10 and pillararenes,7 as well as hydrocarbon molecular belts11 and cage-shaped molecules.12The above strategies utilized symmetric macrocycles or racemic mixtures of ICMs as starting materials. However, since such macrocyclic precursors are predominantly synthesized via condensation of electron-rich aromatic compounds with aldehydes, the majority of classical macrocycles, represented by calixarenes, resorcinarenes, and pillararenes, are composed of electron-rich aromatic segments.13 Consequently, the electronic properties and structural diversity of the macrocyclic cavities remain relatively limited. Recently, our group pioneered a de novo synthetic route for the synthesis of ABCD-type inherently chiral heteracalix[4]aromatics with ee values up to >99%.14–16 Specifically, we first constructed ABCD-type linear oligomers from monomeric fragments through a fragment coupling approach, followed by a Pd-catalyzed asymmetric Buchwald–Hartwig macrocyclization of the linear precursors to synthesize the ICMs (Scheme 1a).14 The de novo synthetic approach enables modular incorporation of monomeric fragments with diverse structural and electronic properties during the synthesis of linear precursors, significantly expanding the structural diversity and functionality of ICMs. We subsequently developed an enantioselective intramolecular Suzuki–Miyaura reaction to synthesize nor-heteracalix[4]arenes featuring a biaryl linkage.16 To further showcase the unique capability of the de novo strategy in accessing unprecedented chiral macrocyclic architectures with tailored functionalities, we present in this work the construction of a novel family of inherently chiral homocalixarenes (Scheme 1b). We propose an intramolecular catalytic asymmetric Sonogashira coupling of linear oligomers bearing terminal alkynes and aryl iodides to access these chiral macrocycles featuring endocyclic alkyne linkages. Post-macrocyclization transformation of the alkyne functionality could enable diverse structural derivatization. The challenges in this process are manifold: not only does it require the construction of a strained endocyclic alkyne, but also the newly formed chemical bond does not dictate the inherent chirality. This stands in sharp contrast to de novo catalytic asymmetric synthesis of axially chiral compounds where chirality is generated directly via the generation of an aryl–aryl bond, facilitating therefore effective chirality transfer between the catalyst and substrates. Notably, the catalytic asymmetric Sonogashira reaction enabling intramolecular macrocyclization with concomitant enantioselective control has never been reported.
 | ||
| Scheme 1 Inherently chiral calixarenes, homooxacalixarenes and nor-heteracalixarenes. | ||
Optimization of reaction conditions
To begin with, our starting reagent linear tetramer 1a containing arylacetylene and aryliodide at both termini can be efficiently synthesized via a fragment coupling approach (SI). We examined the catalytic enantioselective Sonogashira macrocyclization of linear substrate 1a to synthesize ICM 2a. The nature of chiral ligands was found to impact not only the enantioselectivity of the reaction but also the products (Scheme 2 and Table S1). Although bidentate phosphine ligands L1–L3 failed to yield the desired homoheteracalix[4]arene 2a, they unexpectedly afforded the cyclodimer 2a′ (Scheme 2 and SI), albeit in low yields (<5%). The chiral spiro bidentate phosphine ligand SDP (L4) afforded the target homocalix[4]arene 2a in 32% yield, however the product was racemic. Unlike chiral bisphosphine ligands, chiral phosphoramidite (L5–L8) demonstrated superior performance, not only completely suppressing the formation of the dimeric 2a′ but also exhibiting remarkable chiral induction for 2a. As shown in Scheme 2 (see the SI for details), the (R)-1,1′-spirobiindane-7,7′-diol derived phosphoramidites (R)-SIPHOS (L7) stood out as the most promising candidate when compared with BINOL- or H8-BINOL derived ligands (L5 and L6), affording 2a in 30% yield with 46% ee. Notably, the structurally distinct SPHENOL phosphoramidite L8, developed by Sun's group,17 demonstrated further improvements in both yield (36%) and enantioselectivity (66% ee). Using L8 as the chiral ligand, after systematically varying the base, the temperature, the solvent and the palladium sources, the optimum conditions found consisted of performing the intramolecular Sonogashira macrocyclization of 1a in THF/iPrNH (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, c 0.05 M) at 35 °C in the presence of PdI2 (0.1 equiv.), CuI (0.1 equiv.), and L8 (0.2 equiv.). Under these conditions, 2a was isolated in 20% yield with 88% ee. It is noteworthy that the enantiopurity of 2a could be further enhanced to 96% ee via a single recrystallization step using a solvent system of dichloromethane and n-heptane. For a 100 mg scale of 2a, the recrystallization yield was approximately 70–80%.
:1, c 0.05 M) at 35 °C in the presence of PdI2 (0.1 equiv.), CuI (0.1 equiv.), and L8 (0.2 equiv.). Under these conditions, 2a was isolated in 20% yield with 88% ee. It is noteworthy that the enantiopurity of 2a could be further enhanced to 96% ee via a single recrystallization step using a solvent system of dichloromethane and n-heptane. For a 100 mg scale of 2a, the recrystallization yield was approximately 70–80%.
 | ||
| Scheme 2 Enantioselective intramolecular Sonogashira reaction: optimization of conditions. [a] Formation of 2a′ instead of 2a. | ||
Scope of the reaction
With the optimal conditions established, the scope of this enantioselective Sonogashira reaction was next examined. As shown in Scheme 3, varying the substituent at the C6 position of the 1,3,5-triazine from a dimethylamino group to a diethylamino group (2b) or a methoxy group (2c) did not significantly alter the yields and ee of the products. Diminished enantioselectivity was observed when the substituent was further replaced with an ethoxy group (2d). The benzyloxy moieties bearing an electron-donating (Me) or an electron-withdrawing group (F, Cl) at different positions are compatible (2e–2g). However, replacing the bridging CH2 with a carbonyl group prevented the formation of the lactam-containing macrocycle 2h, likely due to restricted conformational flexibility of the linear tetramer 1h. When the amide moiety was relocated from endocyclic to exocyclic, the macrocyclization afforded desired 2i in 30% yield, but with a significantly reduced enantioselectivity (18% ee). This suggests that conformational preference critically determines the success of intramolecular cyclization, particularly for such structurally rigid alkyne-containing macrocycles. Furthermore, although distant from the cyclization site, the acyl group may coordinate to Pd and adversely affect the enantioselectivity. The bridging N–Me could be replaced by an oxygen atom (2j) or N–H (2k), albeit with reduced enantioselectivity in the latter case. However, it is regrettable that the N-allyl group was not compatible under the reaction conditions (2l). The absolute configuration of 2a was assigned to be Ric on the basis of its X-ray structure (Fig. 1).18,19 | ||
| Scheme 3 Enantioselective intramolecular Sonogashira reaction: scope of the reaction. | ||
 | ||
| Fig. 1 Single crystal X-ray molecular structures of Ric-2a (top) and rac-4a (bottom). | ||
To further extend the reaction scope, we examined the O2N1-bridged inherently chiral homocalix[4]arenes. Compared with the previous product 2, one carbon atom was removed from the macrocyclic skeleton. The linear tetramer 3 was synthesized efficiently via a fragment coupling strategy (Scheme 4 and SI). Next, we explored the enantioselective macrocyclization of 3a. Interestingly, while chiral phosphoramide ligands worked well for the intramolecular Sonogashira coupling of 1, they failed with substrate 3a. However, the chiral bisphosphine ligands, previously observed to promote dimerization in the reaction of 1, proved effective here, yielding the desired ICM 4a with good enantioselectivity (Scheme 4 and Table S2). Through condition screening, we found the enantioselective macrocyclization of 3a required elevated temperatures, with C3-TUNEPHOS (L3) emerging as the optimal chiral ligand (see the SI for details). The optimum conditions found consisted of heating a solution of 3a in 1,4-dioxane/iPrNH (v/v = 1:1, c 0.005 M) at 85 °C in the presence of Pd(dba)2 (0.2 equiv.), CuI (0.2 equiv.) and L3 (0.4 equiv.). Under these conditions, compound 4a was isolated in 15% yield with 88% ee. Altering either the dimethylamino group on the triazine ring (4b) or benzyloxy group on the phenyl ring (4c) did not influence the reaction, yielding the products in 18% and 12% isolated yields with 90% and 88% ee, respectively. Overall, linear tetramer 1 afforded the macrocyclization product 2 in 20–30% yield, whereas its shortened analogue 3 (lacking one backbone carbon) gave a lower yield of 10–20% yield. The modest yields were mainly due to competing oligomerizations.
 | ||
| Scheme 4 Catalytic enantioselective Sonogashira macrocyclization of 3. | ||
Structures and applications
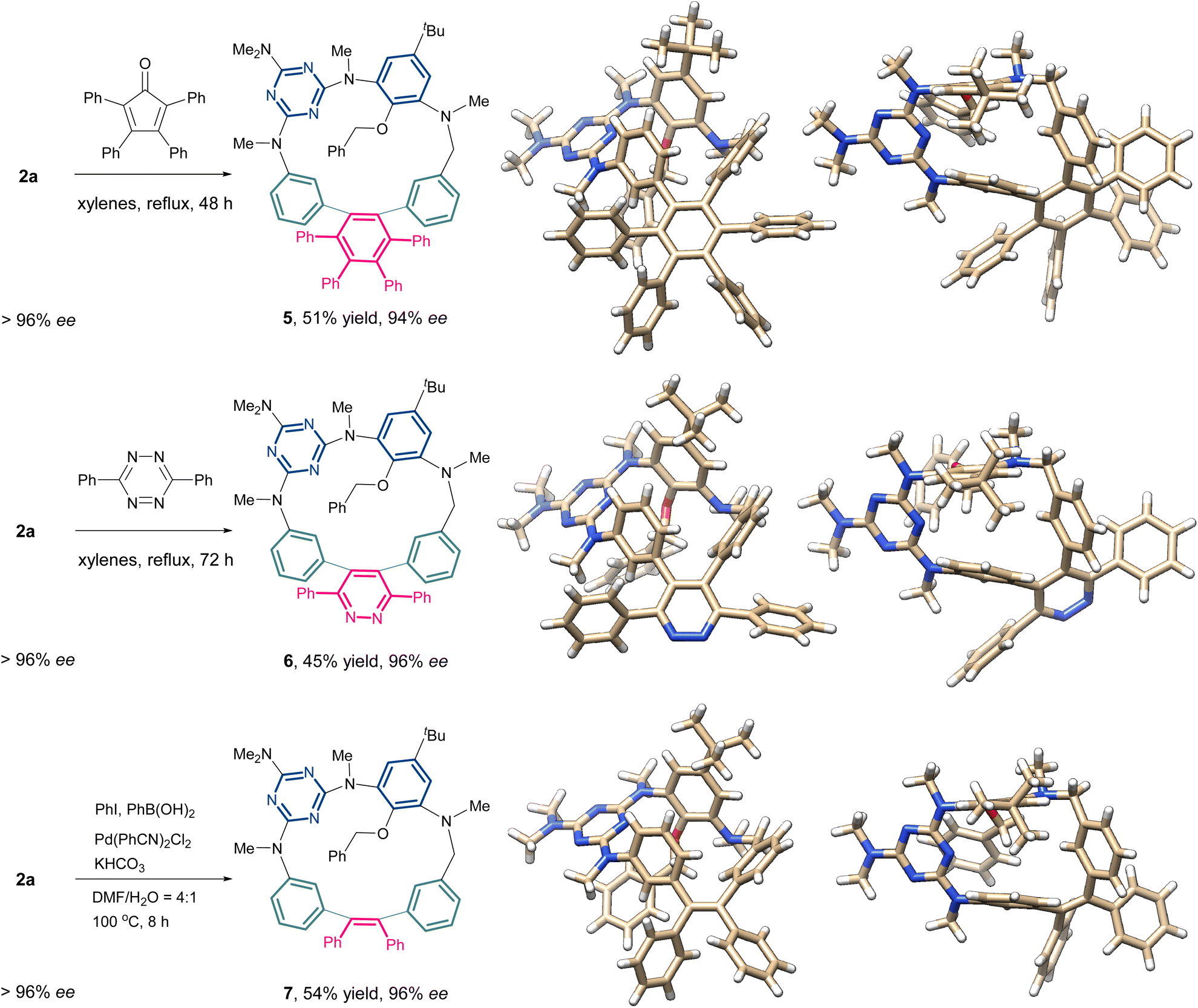
The incorporation of a rigid 1,2-diphenylethyne fragment significantly enhances the structural rigidity and ring strain of the inherently chiral macrocycles. As shown in Fig. 1, the X-ray crystallographic analysis of compounds 2a and 4a demonstrates a striking conformational contrast to conventional heteroatom-bridged calix[4]arenes, which typically adopt the 1,3-alternate conformation.20,21 These homoheterocalix[4]arene macrocycles exhibit a pronouncedly distorted architecture. Apart from the benzene ring bearing sterically hindered substituents at both upper and lower rims, the remaining three aromatic components maintain a nearly coplanar equatorial arrangement. Specifically, 2a adopts a twisted partial cone conformation, while 4a exhibits a distorted 1,2-alternate conformation. The highly rigid macrocyclic framework effectively restricts the ring-flipping of the benzene unit bearing substituents on both upper and lower rims, thus preserving their stable, non-racemizing chiral configurations. Notably, the alkyne bonds within the macrocycles exhibit significant bending (highlighted by bold green lines in Fig. 1). The alkyne bond angles in the 18-membered macrocycle 2a are 173° and 165°, respectively, while the bending is more pronounced in the 17-membered ring 4a, with its alkyne bond angles further compressed to 168° and 157°.The introduction of a strained alkyne moiety within the macrocyclic skeleton offers a versatile handle for post-functionalization of the chiral homocalix[4]arenes, allowing efficient access to a diverse array of novel inherently chiral macrocycles with high optical purity through subsequent alkyne transformation reactions. As illustrated in Scheme 5, using 2a with high optical purity (ee > 96%) as a starting material, macrocycle-to-macrocycle synthesis was achieved. Heating 2a with tetraphenylcyclophentadienone in refluxing xylenes triggered a Diels–Alder/cheletropic reaction sequence, delivering the hexaphenylbenzene-incorporating ICM 5 in 51% yield. Under identical reaction conditions, the cycloaddition of 2a with 3,6-diphenyl-1,2,4,5-tetrazine afforded the ICM 6 embedded with a pyridazine moiety. Additionally, the 1,2-diarylation of the internal alkyne in 2a was achieved via an intermolecular cross-coupling reaction involving an aryl iodide and an arylboronic acid. This transformation produced the desired novel macrocycle 7 in 54% yield, successfully embedding a tetraphenylethene (TPE) unit into the inherently chiral macrocyclic skeleton. The ICMs (5-7) were determined unambiguously with the single crystal X-ray diffraction method (Scheme 5). These novel macrocyclic molecules retain the partial cone conformation of the precursor 2a. Each monomer unit retains its original spatial orientation relative to the macrocyclic equatorial plane throughout the transformation. However, compared to 2a, macrocycles 5–7 exhibit significantly reduced conformational distortion due to the change in hybridization state of the skeletal carbon atoms (from sp to sp2). It is worth noting that the ee was well preserved in the resulting chiral macrocycles (5–7) generated from the above transformations. Prolonged heating under high temperature (with xylenes refluxed for 72 h) did not cause racemization for these 18-membered macrocycles, indicating the high stability of their inherent chirality due to the steric effect of bulky substituents on the benzene unit.
 | ||
| Scheme 5 Construction of novel inherently chiral macrocycles with high optical purity via post-modification of Ric-2a. | ||
Our interest in the chiroptical properties of inherently chiral macrocycles8b,11,14,16,22,23 prompted us to investigate circular dichroism (CD) and circularly polarized luminescence (CPL) of the homoheterocalix[4]arene 2a and its derivatives 5–7. The Cotton effects observed in CD spectra align with their UV-vis absorption maxima (Fig. 2a and c). For compound 2a containing an internal alkyne, the longest-wavelength absorption peak in the UV-vis spectrum is λ = 305 nm, with a molar extinction coefficient (ε) of 1.9 × 104 M−1 cm−1. Correspondingly, the CD spectrum of 2a exhibited a negative Cotton effect at the same wavelength (gabs = −1 × 10−3). Interestingly, despite being derived from the same chiral macrocycle Ric-2a, 5–7 display significantly different Cotton effects at the longest wavelength with sign inversion from negative to positive. Their absorption profiles exhibited substituent-dependent shifts relative to 2a. Introduction of the hexaphenylbenzene moiety in 5 induced a pronounced hypsochromic shift and sign inversion (Δλ = −23 nm, gabs = +2.3 × 10−3), while incorporation of either pyridazine (6) or TPE (7) fragments resulted in bathochromic shifts of Δλ = +43 nm (gabs = −2.5 × 10−3) and +21 nm (gabs = +4.6 × 10−3), respectively. X-ray crystallographic analysis revealed that the observed spectral shifts correlate with distinct conformational changes induced by each substituent (Scheme 5 and Fig. S22–S27). Upon irradiation, compound 2a in CH2Cl2 solution exhibited fluorescence emission at λem = 454 nm with a quantum yield (Φ) of 3.0%. In contrast, 5 displayed significantly quenched emission (λem = 454 nm, hypsochromically relative to 2a), while macrocycles 6 and 7 were essentially non-emissive in solution (Fig. 2d and the SI). Moreover, we found that the single crystals of compound 7 emit cyan fluorescence under UV irradiation, whereas crystals of 5, 6, and 2a remain non-emissive (Fig. 2b). The fluorescence emission wavelength of 7 in the solid state was at 480 nm, bathochromically shifted relative to 2a and 7 in CH2Cl2 solution (Fig. 2d). Single crystal packing analysis of 7 revealed that the phenyl groups of the TPE moiety participate in multiple intermolecular CH-pi interactions with neighboring macrocycles in the solid state (Fig. S27, SI). This rigidification effectively suppresses the free rotation of the TPE's exocyclic phenyl moieties, resulting in aggregation induced emission (AIE) characteristics. Accordingly, compound 7 derived from Ric-2a exhibited no detectable CPL in solution. However, it displayed a pronounced positive CPL signal with a luminescence dissymmetry factor (glum) of 1.5 × 10−3 (Fig. 2b) in the solid state, suggesting its potential in the area of chiral photofunctional materials.
 | ||
| Fig. 2 (a) CD spectra of 2a, 5, 6, and 7 in CH2Cl2 (c 1 × 10−5); (b) CPL spectrum of 7 in the solid state and the crystal photos of compound 7 under daylight and UV light; Normalized UV-vis (c) and fluorescence (d) spectra of 2a, 5, 6, and 7 in CH2Cl2 (c 2 × 10−5) and 7 in the solid state. | ||
Conclusions
In summary, we have designed and synthesized a new class of enantioenriched homo-heteracalixaromatics by means of de novo macrocyclization using an intramolecular Sonogashira cross-coupling reaction. Remarkably, the ICMs synthesized display distinct structural and functional features compared to conventional calixarenes and heteracalixarenes. The strategic integration of strained alkyne motifs within macrocyclic frameworks facilitates efficient post-functionalization, enabling the construction of an expanded library of inherently chiral macroarchitectures with unprecedented structural diversity. Additionally, we have revealed the structures and intriguing chiroptical properties, including the AIE effect and the pronounced CPL of the acquired ICMs in the solid state. This research opens the door for the exploration of novel and sophisticated inherently chiral macrocyclic structures with outstanding physical and chemical properties, as well as potential applications.Author contributions
Y.-F. J., S. T., J. Z. and M.-X. W. conceived and designed the experiments. Y.-F. J. and Y. Z. carried out the experiments. S. T., J. Z. and M.-X. W. wrote the manuscript. All authors contributed to the reviewing and editing of the manuscript and SI.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2412922 (rac-5), 2412924 (rac-6), 2412925 (rac-7), 2412927 (rac-4a) and 2412928 (Ric-2a) contain the supplementary crystallographic data for this paper.18a–eThe authors declare that data supporting the findings of this study are available within the paper and the supplementary information (SI), as well as from the authors upon request. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc05923b.
Acknowledgements
We are thankful to the National Natural Science Foundation of China (No. 22171160, 22371161, 21920102001), EPFL (Switzerland), and Tsinghua University Dushi Program for financial support.Notes and references
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- A. Dalla Cort, L. Mandolini, C. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198–1199 RSC.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- G. E. Arnott, Chem.–Eur. J., 2018, 24, 1744–1754 CrossRef CAS.
- Q.-L. Lu, X.-D. Wang, S. Tong, J. Zhu and M.-X. Wang, ACS Catal., 2024, 14, 5140–5146 CrossRef CAS.
- Y.-Q. Mao, Y. Zhang, S. Tong, J. Zhu and M.-X. Wang, Org. Lett., 2023, 25, 3936–3940 CrossRef CAS PubMed.
- (a) X.-H. Zhou, X. Zhang, Y.-R. Song, L. Xue, L.-T. Bao, W.-T. Xu, X.-Q. Wang, H.-B. Yang and W. Wang, Angew. Chem., Int. Ed., 2025, e202415190 CAS; (b) T.-R. Luan, C. Sun, Y.-L. Tian, Y.-K. Jiang, L.-L. Xi and R.-R. Liu, Nat. Commun., 2025, 16, 2370 CrossRef CAS PubMed; (c) A. Konter, J. Rostoll-Berenguer, C. Besnard, B. Leforestier and C. Mazet, ACS Catal., 2025, 15, 2607–2619 CrossRef CAS.
- For pioneering works, see: (a) J. K. Browne, M. A. McKervey, M. Pitarch, J. A. Russell and J. S. Millership, Tetrahedron Lett., 1998, 39, 1787–1790 CrossRef CAS; (b) X. Zhang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2023, 14, 827–832 RSC; (c) Y.-Z. Zhang, M.-M. Xu, X.-G. Si, J.-L. Hou and Q. Cai, J. Am. Chem. Soc., 2022, 144, 22858–22864 CrossRef CAS PubMed.
- For selected publications, see: (a) Y.-K. Jiang, Y.-L. Tian, J. Feng, H. Zhang, L. Wang, W.-A. Yang, X.-D. Xu and R.-R. Liu, Angew. Chem., Int. Ed., 2024, e202407752 CAS; (b) S. Yu, M. Yuan, W. Xie, Z. Ye, T. Qin, N. Yu and X. Yang, Angew. Chem., Int. Ed., 2024, e202410628 CAS; (c) P.-F. Qian, G. Zhou, J.-H. Hu, B.-J. Wang, A.-L. Jiang, T. Zhou, W.-K. Yuan, Q.-J. Yao, J.-H. Chen, K.-X. Kong and B.-F. Shi, Angew. Chem., Int. Ed., 2024, e202412459 CAS; (d) X.-Y. Zhang, D. Zhu, R.-F. Cao, Y.-X. Huo, T.-M. Ding and Z.-M. Chen, Nat. Commun., 2024, 15, 9929 CrossRef CAS; (e) V. Dočekal, L. Lóška, A. Kurčina, I. Císařová and J. Veselý, Nat. Commun., 2025, 16, 4443 CrossRef.
- H. Han, X.-G. Wang, S. Tong, J. Zhu and M.-X. Wang, ACS Catal., 2025, 15, 6018–6024 CrossRef CAS.
- X.-Y. Wang, X.-Y. Zhang, S. Tong, Q.-H. Guo, M.-L. Tan, C.-J. Li and M.-X. Wang, CCS Chem., 2024, 6, 1198–1210 CrossRef CAS.
- S. Fang, Z. Bao, Z. Liu, Z. Wu, J.-P. Tan, X. Wei, B. Li and T. Wang, Angew. Chem., Int. Ed., 2024, e202411889 CAS.
- P. Neri, J. L. Sessler and M.-X. Wang, in Calixarenes and Beyond, Springer International Publishing, Cham. 2016 Search PubMed.
- S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed.
- X.-C. Li, Y. Cheng, X.-D. Wang, S. Tong and M.-X. Wang, Chem. Sci., 2024, 15, 3610–3615 RSC.
- Y.-F. Jiang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2024, 15, 12517–12522 RSC.
- R. Zhang, S. Ge and J. Sun, J. Am. Chem. Soc., 2021, 143, 12445–12449 CrossRef CAS PubMed.
- (a) CCDC 2412922: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lzv6n; (b) CCDC 2412924: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lzv8q; (c) CCDC 2412925: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lzv9r; (d) CCDC 2412927: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lzvct; (e) CCDC 2412928: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2lzvdv.
- Note: regarding the assignment of the absolute configuration, details are provided in the SI. To assign the absolute configuration of the inherently chiral macrocycles, we adopt the nomenclature rules for planar chirality, selecting the highest-priority triazine ring as the principal plane. Thus, the absolute configuration of compound 2a in the article is assigned as Ric, where the subscript “ic” stands for inherent chirality..
- J.-T. Li, L.-X. Wang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2014, 79, 2178–2188 CrossRef CAS.
- M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- J.-H. Chen, Z.-Y. Jiang, H. Xiao, S. Tong, T.-H. Shi, J. Zhu and M.-X. Wang, Angew. Chem., Int. Ed., 2023, e202301782 CAS.
- Y. Peng, S. Tong, Y. Zhang and M.-X. Wang, Angew. Chem., Int. Ed., 2023, e202302646 CAS.
| This journal is © The Royal Society of Chemistry 2026 |