
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDigitized dataset of aqueous acid dissociation constants
Jonathan W. Zheng ,
Olivier Lafontant-Joseph and
William H. Green*
,
Olivier Lafontant-Joseph and
William H. Green*
Massachusetts Institute of Technology, Department of Chemical Engineering, USA. E-mail: whgreen@mit.edu
First published on 24th April 2026
Abstract
The acid dissociation constant (pKa) quantifies the acidity of a compound, which is crucial for applications including drug design, environmental fate studies, and chemical synthesis. However, high-quality open-source digital pKa datasets are scarce, which limits the ability for researchers to search for properties of individual compounds, while also limiting the potential of data-driven predictive models. In this work, we release the IUPAC Digitized pKa Dataset, a digital version of a critically-assessed collection of data compiled up to 1970. The dataset includes metadata such as temperature, measurement method, assessed reliability of data, and chemical identifiers such as SMILES and InChI strings. The dataset spans 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 222 entries across 10564 unique molecules, making it the largest FAIR open-source dataset publicly available for aqueous pKa data. Herein, we detail the data digitization and checking process, and assess the informational space spanned by the data. We compare the new digital dataset to other widely-used datasets. Several pKa predictors have been trained using these other datasets, but often have not been reliably tested due to overlap between the training and test data. We use the data to train a macroscopic pKa predictor and determine its accuracy using overlap-free test data. The full dataset is available at https://doi.org/10.5281/zenodo.7236452, and the models and data splits used in this study are available at https://doi.org/10.5281/zenodo.18165948.
222 entries across 10564 unique molecules, making it the largest FAIR open-source dataset publicly available for aqueous pKa data. Herein, we detail the data digitization and checking process, and assess the informational space spanned by the data. We compare the new digital dataset to other widely-used datasets. Several pKa predictors have been trained using these other datasets, but often have not been reliably tested due to overlap between the training and test data. We use the data to train a macroscopic pKa predictor and determine its accuracy using overlap-free test data. The full dataset is available at https://doi.org/10.5281/zenodo.7236452, and the models and data splits used in this study are available at https://doi.org/10.5281/zenodo.18165948.
Introduction
The acid dissociation constant (also known as pKa) is an important property involved in pharmacokinetics, environmental fate, chemical manufacturing, organic synthesis, analytical chemistry, thermodynamics, and numerous other applications.1–11 pKa is defined from the equilibrium constant of the acid dissociation reaction for species AH in a solvent as:| AH(solv.) ⇌ H+(solv.) + A−(solv.) | (1) |
 | (2) |
| pKa = −log10(Ka) | (3) |
Under this definition, pKa strictly refers to proton loss. The International Union of Pure and Applied Chemistry (IUPAC) prefers to call the cation H+ a “hydron” rather than a proton, as the term encompasses such isotopes as the deuteron.13 In this work, we will use the term “proton” interchangeably with “hydron” due to its wider current acceptance.
A common way of representing proton gain is by reporting the pKa of its conjugate acid, a term sometimes called the “basic pKa”. In this convention, the “basic pKa” of compound B numerically represents the acidic dissociation constant for BH+, the conjugate acid of B, or:
| BH+(solv.) ⇌ B(solv.) + H+(solv.) | (4) |
This is sometimes confusingly referred to as the “pKa of B”, with the understanding that the value refers to the conjugate acid's acidity since B itself is basic. To avoid ambiguity in this work, we use the convention that the pKa of a species strictly refers to its acidic dissociation, as in eqn (1). We use the term pKaH(B) (as recently recommended by IUPAC)14 to designate the pKa corresponding to the conjugate acid, i.e. pKa(BH+).
Often, AH contains several ionizable protic sites, and there might be several isomers of HA in equilibrium, such as zwitterions and uncharged protomers. The macroscopic pKa corresponds to the equilibrium constant for an overall charge transition, so the species A− in eqn (1) is an equilibrium mixture of several isomers with protons bonded to different atoms. In contrast, the microscopic pKa refers to the acidity of a specific microstate (corresponding to a specific isomer of AH losing a proton at a specific ionization center to form a specific isomer of A−). Whereas the former is most commonly measured, e.g. by measuring the pH at equilibrium after half of the initial AH has been deprotonated, the latter is more readily obtainable using simulations. A macroscopic pKa can be computed from the microscopic pKa values by Boltzmann-weighting the respective microstates.15–17 For monoprotic acids without tautomers, the macroscopic and microscopic pKa values are the same. For polyprotic acids with large separation between pKa values and no tautomeric effects, they are also approximately the same. In all other cases, the macroscopic and microscopic pKa can be different. Most data in this compilation are macroscopic pKa values, and do not include any information about different acidity centers.
Owing to the importance of pKa in numerous applications, considerable effort has been made to accurately predict pKa values across a range of solvents, primarily in water. Early methods typically involved group-based methods, in which the pKa of acids are assigned based on linear free energy corrections, and often depend on specific moieties. Popular examples include the Hammett equations (for benzoic acid derivatives) and Taft equations (which estimates pKa effects of adding groups to a parent compound).18
Deep learning has recently emerged as a tool for quick and accurate prediction of chemical properties.19–25 However, large collections of data are required to tune large numbers of parameters in these models. Recently, numerous pKa prediction models leveraging deep learning have been developed and made publicly available.11,26–33 These models are trained on open-source datasets, which include large quantities of pKa data, and sometimes also calculations (or “synthetic data”) to augment the experimental data.31–34 Despite the apparent good performance of the models, low data quantity and quality remain major obstacles. Furthermore, though many recent efforts claim to predict microscopic pKa values, they are actually trained on macroscopic pKa data. A contributor to this issue is that many datasets do not list the measurement method or type of pKa, and so this information is not immediately obvious to the modelers. Several additional data-related issues are listed below.
Limitations of existing datasets
Several datasets are currently utilized in machine learning applications or as reference sources, but have issues in data quality, quantity, provenance, reported conditions, and other issues.ChEMBL
The ChEMBL database35–37 is a widely-used26,32,34,38 biochemical data repository. Among numerous other data such as bioactivity, it also includes approximately 3 million pKa1 and pKaH1 (strongest proton loss and proton gain values, respectively) for about 2 million molecules, computed using the ChemAxon Protonation tool (Marvin).39 The values appear to correspond to ChemAxon calculations in the macroscopic static pKa mode at 0.1 M ionic strength (“static” means that charged forms of input molecules are converted to neutral forms prior to calculation).40,41Because the data are computed rather than experimental, and owing to the very large size of the data, they have been typically used for pretraining (though sometimes used exclusively for training). However, the data has been shown to include some incorrect values, and commonly are misused in modeling studies due to a misinterpretation of the meaning of “acidity” and “basicity” for amphiprotic compounds.40
Though widely-used and by far the largest set of values, it originates from computations, which limits the predictive power of models to the error of the calculations. Each datapoint is provided without an estimate of uncertainty, making it difficult to know which values are accurate. Also, only pKaH1 and pKa1 are reported, which limits the ability to model polyprotic compounds.
iBonD
The iBonD database was developed by Prof. Jin-Pei Cheng and collaborators at Tsinghua University. It is comprised of more than 40000 pKa entries across a variety of solvents, compiled from existing academic literature. It is widely used for manual search as a reference source as well as in machine learning.42
The recent work by An et al.,43 Luo et al.,32 and Nevolianis & Zheng et al.,11 have separately transformed portions of the iBonD data into tabulated forms more convenient for data science applications. The data have been used in several machine learning models.
The data entries are reported to have undergone individual curation and evaluation, and a single acidity center for each dissociation is reported. However, metadata such as temperature and assessed reliability of data are not reported. Additionally, some errors in non-aqueous data (due, for instance, to different choices of energy scales) are present.11
DataWarrior
The DataWarrior software suite44 includes a compilation of pKa data with unknown provenance. Some recent models were trained on this data,27 which includes close to 10000 entries. Due to the lack of provenance and uncertainty estimates, the quality of the data are unclear.
Other datasets not analyzed in this work
A few other datasets have been used to train models. QSAR Toolbox45 is a software that includes approximately 30000 pKa entries. The quality of the data is unclear, and distinctions are not always made between “acidic” and “basic” pKa values, so additional processing may be required; nevertheless, the data have been used for modeling.30
Another potential data source is crowdsourced data, such as in Online Chemical Modeling Environment (OCHEM), which includes provenance but includes inconsistent, mixed-quality metadata, thereby requiring additional processing before usage.46
Other data are proprietary and hence not applicable for open-source research. The largest described collections of pKa data are held by companies: for instance, the S + pKa model was trained on 70669 datapoints that combined information from Bayer Pharma, Roche, Genentech, and Bayer CropScience.47,48
Overall challenges
Despite their issues, the datasets mentioned above are frequently used as training or reference data. Sometimes, several of these datasets are combined, with duplicate values removed on the basis of prioritizing data from more reliable sources.27,30 But the overall quality of available digitized pKa data, including provenance and metadata, remains low.Hence, there is a need for high-quality data to use for pKa prediction, as well as further clarity on how these datasets relate to one another.
Aims of this work
Herein, we introduce a large, high-quality dataset of acid dissociation constants with provenance of all original sources. The dataset includes 24222 entries spanning 10564 unique compounds, with pKa values spanning up to six proton gains and six proton losses. The dataset includes rich metadata not included in any other compilation, such as temperature, pressure, assessed reliability, and measurement method, as well as chemical identifiers SMILES and InChI strings.
We discuss the intended use cases for this data: as a reference source, and for machine learning. We compare this dataset to the iBonD and DataWarrior collections. We show that all commonly-used pKa datasets (including this one) include data overlaps with common benchmarks, requiring data pruning for fair comparison. Finally, we analyze macroscopic pKa models trained on variants of this data and the ChEMBL database using Chemprop.49
Dataset background
This article details the digitization and curation of data presented in three reference books published by the International Union of Pure and Applied Chemistry (IUPAC):(1) Serjeant: ionisation constants of organic acids in aqueous solution; E. P. Serjeant and Boyd Dempsey; Oxford/Pergamon (1979) (Oxford IUPAC chemical data series).50
(2) Perrin: dissociation constants of organic bases in aqueous solution; D. D. Perrin; Butterworths (1965).51
(3) Perrin Supplement: dissociation constants of organic bases in aqueous solution, Supplement 1972; D. D. Perrin; Butterworths (1972).52
IUPAC provided written permission to scan and digitize the data, provided that the output data are reviewed by IUPAC, posted in an IUPAC-owned repository, and support the FAIR (Findable, Accessible, Interoperable, Reusable) data principles. With their permission, we scanned and digitized the reference books. A digitized copy of Perrin (1965) was obtained from the Internet Archive with IUPAC's permission.
A commercial version of these data sources is separately available under OpenEye Scientific Software.53 That collection was parsed independently of this collection and includes additional database features such as tautomer enumeration, as well as the aqueous data from Kortum54 and the non-aqueous data from Izutsu.55 The number of data in that collection is slightly different than in this work, as we were unable to resolve the structures of some compounds and thereby did not report them.
This work is a digitized adaptation developed from IUPAC source data with permission. We emphasize that this manuscript should not be considered an official IUPAC technical report (which would be published in the journal Pure and Applied Chemistry). We make no guarantees on the faithfulness of the digitization process to the print source, or for strict adherence to IUPAC conventions.
The data are publicly available at https://doi.org/10.5281/zenodo.19112621. At the time of publication, the version of the dataset is 2.3d, and the data are provided under a CC BY-NC 4.0 license.
Methods
Dataset construction through digitization
A general discussion of the workflow is depicted in Fig. 1 and described below: | ||
| Fig. 1 Visualization of digitization workflow. Images were converted to text via OCR, and then processed with various cheminformatics workflows. | ||
(1) Digital scans of the reference books were obtained. With IUPAC's permission, the reference books were scanned by the authors in the MIT Libraries, or a digital copy was obtain from the Internet Archive.
(2) Optical character recognition (OCR) was employed to convert the scanned images into text, ordering the data into tables using Amazon Textract.56 Information from these tables were further parsed and processed into categories of data, e.g. pKa type, chemical name, reference, method used, and so on.
(3) The IUPAC names of the compounds were translated into SMILES and InChI strings using OPSIN,57 ChemAxon molconvert,39 PubChem,58–60 and the Chemical Identifier Resolver.61 SMILES strings were accepted only if the same string was unanimously returned by 2+ methods. Of the entries with translations missing, inconsistent, or from only one translation source, we manually parsed the IUPAC names into SMILES strings following IUPAC conventions for naming. We note that tools have recently been developed to expedite and automate this process, such as MoleculeResolver, but were not used in this work.62
(4) The dataset was standardized to reduce the number of unique entries and make the data more amenable to data processing. For instance, “room temperature” was converted to 25 °C, and pressures were parsed into a separate column wherever applicable.
(5) To resolve the pKa type, we followed patterns in the reference books to confidently assign labels for a majority of the entries (for example, in the data compiled by Perrin, pKa data in descending order corresponded to pKaH data, whereas in ascending order corresponded to amphiprotic molecules). For approximately 4000 entries, we could not automatically discern the pKa type and therefore manually assigned them based on their numeric values and chemical structures.
(6) The data were analyzed and visualized, checking also for errors and outliers.
Validation of dataset values
All of the digitization efforts were manually double-checked by human reviewers. We double-checked that the dataset transcriptions were faithful to the original print sources.The data were checked to identify suspicious entries that needed verification or correction, such as entries with abnormally high or low pKa values, missing locants in chemical names, common typos in the metadata, species with high deviation among multiple measurements, and implausible chemical structures.
ChemAxon's Protonation software predictions were checked against pKa values for all of the species. The majority of calculated values agreed with our experimental data within 1 pKa unit, though a few datapoints showed large disagreement, usually due to inconsistencies in acidity type assignment. If deviations exceeding 4 pKa units were observed, we manually reviewed the data in our collection and made corrections if necessary.
We reviewed entries with large deviations for the same acidity type at a given temperature. We also confirmed that pKaH values were lower than pKa values for amphiprotic compounds.
Results and discussion
Dataset layout
The dataset is presented as a .csv with column headers described in Table 1. The headers include chemical identifier information (IUPAC names, SMILES strings, and InChI strings); pKa descriptors (a pK type, numerical value, and acidity label); experimental conditions (temperature, pressure, method, cosolvent); metadata (data provenance, name-to-structure translation methods); and critical assessment (reliability, extra comments).| Column header | Description |
|---|---|
| unique_ID | Identifier corresponding to each unique compound in the corresponding source; note this is unique to the entry rather than to the molecular identifier, as separate sources may contain the same compound |
| SMILES | Isomeric SMILES string canonicalized in rdkit |
| InChI | InChI string corresponding to compound; unique molecular identifiers conducive to look-up |
| pka_type | Type of acid dissociation; e.g. pKa = acid dissociation, pKaH = dissociation of conjugate acid (sometimes called “basic” pKa), pKb = base association |
| pka_value | Numerical value of pKa |
| T | Temperature of measurement (in °C), standardized to a numeric value if possible |
| remarks | Comments about this entry from the print source; e.g. ionic strength, experimental considerations |
| method | Experimental method for this entry |
| assessment | Critical assessment of source's reliability. The original authors (Perrin, Serjeant) assessed errors in pKa as: ≤0.005 for “reliable” entries, ≤0.04 for “approximate”, >0.04 for “uncertain”, and high errors for “very uncertain” |
| ref | Code corresponding to citation for original source of the data |
| ref_remarks | Additional comments that apply to all entries with the “unique_ID” with the same reference |
| entry_remarks | Additional comments that apply to all entries with this “unique_ID” |
| original_IUPAC_names | IUPAC names from the original print sources |
| name_contributors | Method(s) of obtaining SMILES strings from IUPAC names |
| num_name_contributors | Number of methods used to obtain SMILES strings from IUPAC names |
| original_IUPAC_nicknames | If applicable, secondary/common names that were also supplied in print source |
| source | Source book (Serjeant, Perrin, or Perrin Supplement; see main text) |
| pressure | If available, pressure of the measurement (units typically of atm or bar) |
| acidity_label | “A” for acidic, “AH” for conjugate acid, “B” for basic, or “other” |
| original_T | Original unprocessed temperature from the print source |
| cosolvent | Cosolvent information parsed from the entry |
We intend for this dataset to be useful for both experimental and machine learning applications. We intend that the InChI strings will allow users to look-up chemicals by a unique ID. From a computational perspective, we hope that the SMILES strings will aid the development of machine learning models and quantum chemical workflows, since many software packages requires SMILES strings as inputs.
A terminological area of note is the naming of “acidic” and “basic” pKa values for compounds that form zwitterions in solution. Historically, an ampholyte such as glycine often has its lower pKa labeled as an acidic pKa and its higher pKa as a basic one; however, such ordering is inconsistent with the microstates of the relevant protomers undergoing dissociation. Such labels have led to serious confusion in model development in recent years. For this reason, although the 3 source books used the historical “acidic” and “basic” naming precedent, we have in this work elected to use pKaH and pKa terminology, which has recently been recommended by IUPAC.14 For further discussion about potential confusion with pKa terminology, we refer interested readers to our previous work on this topic40 and to recent literature.63
Dataset contents
This dataset includes 24222 entries spanning 10564 unique chemical species. Most compounds include multiple pKa values with the same dissociation type but at different experimental conditions. Considering each pKa type as unique regardless of experimental condition, there are 14681 such entries, reflecting the polyprotic and amphoteric nature of some of the 10564 species.
The compounds are mostly small organic molecules, centered around 10 heavy atoms, with a right tail indicating some larger molecules including a few drug compounds (Fig. 2). Most of the molecules have at least one site that can accept a proton (Fig. 2c), and many compounds include at least one hydrogen bond donor site (Fig. 2d).
 | ||
| Fig. 2 Summary dataset distributions, including: (a) number of heavy atoms, (b) molar mass, (c) calculated hydrogen bond acceptors, and (d) calculated hydrogen bond donors. The latter two properties were computed using rdkit v2024.03.1. Darker color represents higher density of points. | ||
The distributions of pKa1 and pKaH1 shown in Fig. 3 both have peaks around 4 and 9 pKa units. The majority of pKa1 values fall in the range of 3 to 11, representing weak-to-medium acids and bases. But there are a significant number of data on very weak bases (pKaH1 < 3) and very weak acids (pKa1 > 11).
 | ||
| Fig. 3 First acidic and basic pKa values in the dataset, across all species, at approximately 25 °C. For acids with multiple values near room temperature, one value between 20–30 °C was reported. Both distributions exhibit peaks at around 4 and 9 pH units, while the basic dissociation shows a long left-skewed tail. Overlapping bins are shown in purple. | ||
A large variety of pKa types is present in this dataset. The data include both pKaH and pKa up to six charge states (Fig. 4). Most entries are related to first and second dissociations. Just one compound includes a sixth proton gain pKaH, whereas around ten include a sixth proton loss pKa.
 | ||
| Fig. 4 Distribution of pKa types across all compounds and temperatures in the dataset (log-y scale). Most entries are for first and second dissociations, with slightly more first basic than first acidic pKa values. Darker color represents higher density of points. | ||
pKa data are recorded across a number of conditions, including temperature, pressure, experimental method, and evaluated reliability.
Most entries reported herein were measured near or at room temperature (Fig. 5). A small number of entries are measured or calculated at elevated temperatures, allowing the temperature dependence of pKa to be visualized or estimated.
 | ||
| Fig. 5 Temperature distribution for all entries in the dataset (logy-scale). The vast majority of entries are at room temperature, although a few entries are recorded as high as 225 °C. Values above 100 °C were either obtained at elevated pressures, or computed from temperature-based extrapolations at 1 bar. Darker color represents higher density of points. | ||
Ionic strength is sometimes also recorded. For most applications of pKa, it is assumed that the ionic strength is near zero. Most of the ionic strength measurements in this dataset are low. The ionic strength is presented in many different formats; for example, either directly as ionic strength I, as c in molar concentration, as m in concentration per kilogram of water, or as κ in specific conductance of water (in 10−6 ohm−1 cm−1). In many cases the value provided is approximate, or only a range is given. At the time of publication, the ionic strength is included in the remarks column, rather than parsed in a separate column.
The vast majority of measured data in this compilation are from electrochemical or optical methods (Fig. 6), which are only capable of discerning macroscopic pKa values. Only 30 measurements were made with NMR, which can provide information about the microstates. Hence, this dataset is intended to be used as a macroscopic pKa database, though it is in principle possible to employ physical chemical information (such as through quantum chemical simulations) to decompose the macroscopic values into the corresponding microstates.
 | ||
| Fig. 6 Measurement methods employed for each datapoint. The majority of measurements were obtained using electrometric experiments. Darker color represents higher density of points. | ||
Table 2 shows the details of the original print sources. The works by Perrin were focused largely on basic (pKaH) values, whereas that of Serjeant was focused on acidic values. Notably, acidic pKa values prior to 1961 are missing – those were compiled by Kortum et al.54 for 1056 compounds, and published only in German. The Kortum work contains values for aliphatic and alicyclic carboxylic acids, aromatic carboxylic acids, phenolic acids, and other acids including phosphoric acid ethers, and sulfonic, phosphonic, and phosphinic acids. We unfortunately were unable to translate the Kortum work with high confidence, and hence it is not included herein. We hope to incorporate this data in the future.
| Reference source | # Entries | # Molecules | Description |
|---|---|---|---|
| perrin | 7769 | 3433 | Values mostly for organic bases, collected up to 196151 |
| perrin_supp | 7147 | 3914 | An add-on to the first Perrin compilation, collected from 1961 to 1970, mostly of values for organic bases. Includes some corrections to Perrin, which were incorporated in this digitization52 |
| serjeant | 9295 | 4108 | Supplement to work by Kortum et al.54 Data collected from 1961 to 1970, mostly of acidic dissociations50 |
Representative data examples
Herein, we present representative examples that underscore the breadth of the available data and metadata. | ||
| Fig. 7 Isobars for acetic acid pKa varying across temperature. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety, which forms a resonance-stabilized cation upon protonation.
N moiety, which forms a resonance-stabilized cation upon protonation.
| pKa1 | Acidic molecule | pKaH1 | Basic molecule |
|---|---|---|---|
| −6.4 |  |
14.1 |  |
| −5.8 |  |
14.0 |  |
| −5.2 |  |
14.0 | 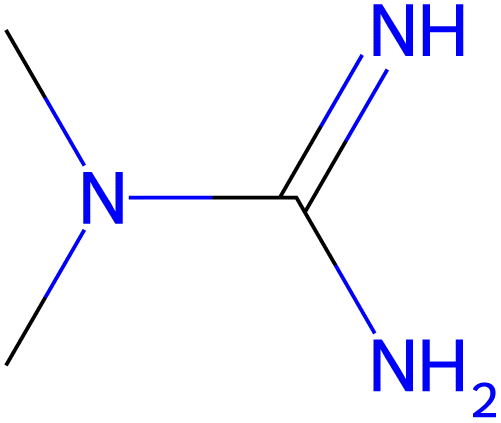 |
| −3.9 | 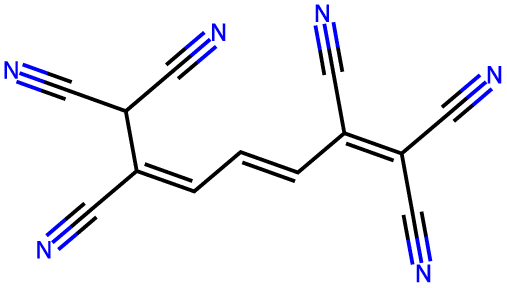 |
14.0 | 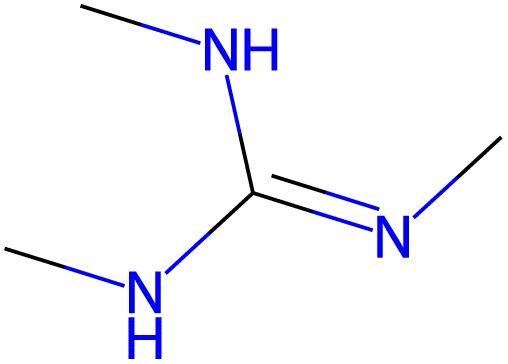 |
| −3.5 | 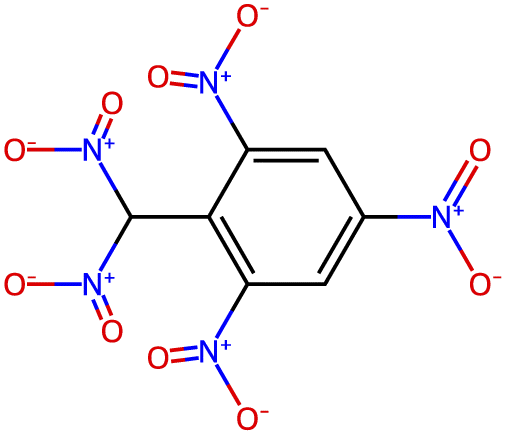 |
13.9 | 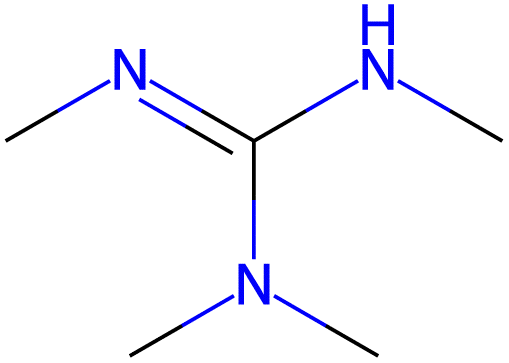 |
| −3.0 | 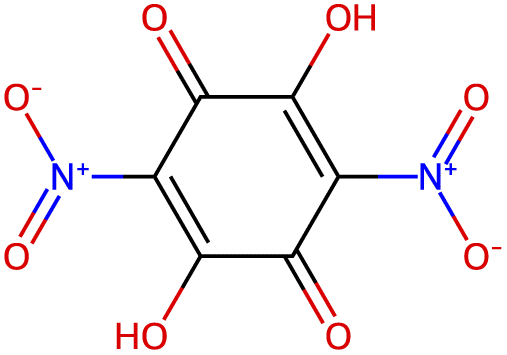 |
13.9 | 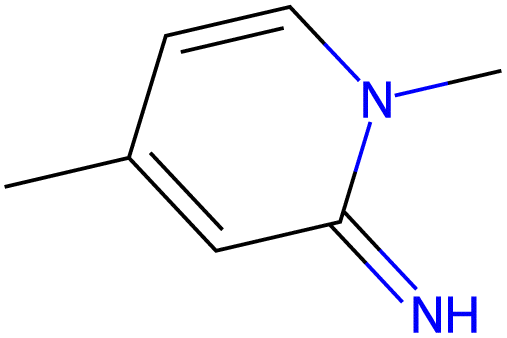 |
| −2.8 | 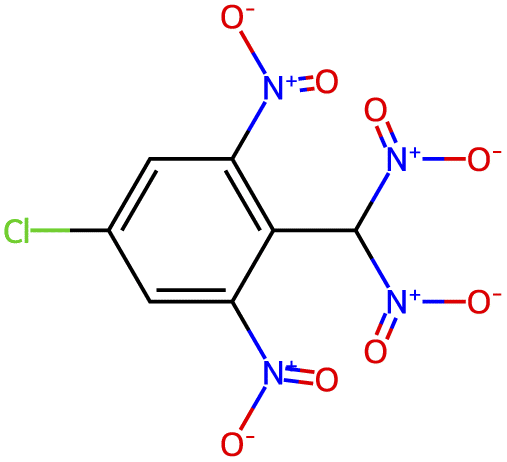 |
13.9 | 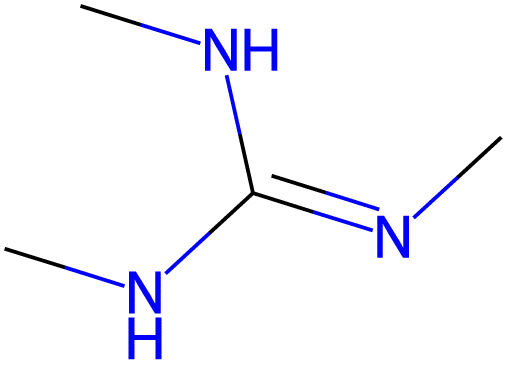 |
| −2.8 | 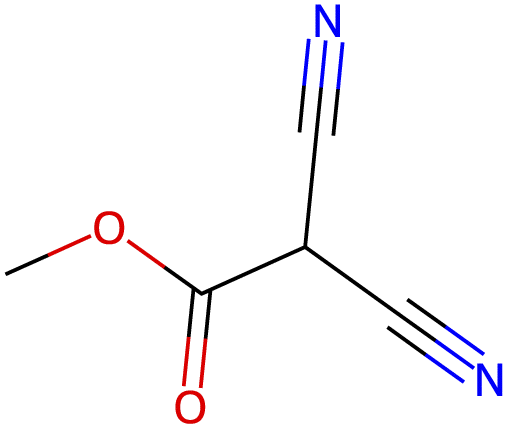 |
13.9 | 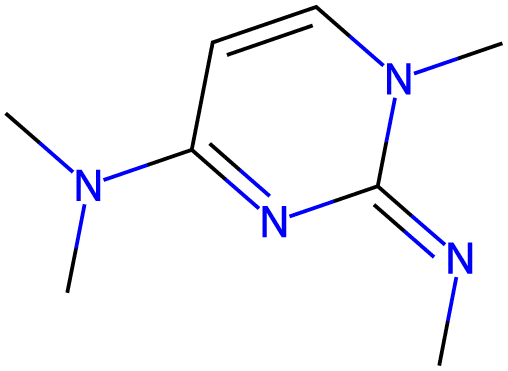 |
Usage as reference data
We intend that the data should be used as reliable reference data. We aim to follow FAIR (Findability, Accessibility, Interoperability, and Reuse) principles to allow users to readily, freely access the data.• Findability – each compound has a unique identifier. Rich metadata are provided, with descriptions that explicitly describe the type of data.
• Accessibility – the data and metadata can be accessed freely (through free download) – currently through Zenodo – in a .csv file.
• Interoperability – the .csv format with chemical identifiers allows it to be readily used in cheminformatics workflows and beyond.
• Reusability – license information is provided in the README included in the download repository, and sources are given for each datapoint.
Because the data is released in a digital format, it is also readily updateable. Errors in the database can be pointed out by users and subsequently fixed in the datasheet.
Comparison to other datasets
We compared the chemical diversity and pKa values of the species in this dataset to those of data in other commonly-used pKa datasets in machine learning. | ||
| Fig. 8 Comparison of unique compounds in three large datasets: this dataset (top-left), DataWarrior (top-right), and iBonD aqueous values (bottom). Unique compounds were determined by identifying intersections of sanitized InChI strings among the datasets. | ||
Fig. 9 shows the chemical space spanned by the compounds, represented by a UMAP plot.64 The domain is fairly consistently covered among the IUPAC, DataWarrior, and iBonD datasets, with overlapping points spanning practically all regions of this UMAP plot. That said, there are regions of chemical space where more examples are present for certain datasets versus the others. In particular, the bottom-right region contains mostly IUPAC data, which correspond mostly to simple nitrogen-containing heterocyclic compounds (such as pteridine and pyrimidine derivatives). In contrast, the top-center, top-left, and bottommost regions of the UMAP plot are somewhat more heavily populated by the aqueous iBonD data, which correspond to sulfonamides and large, druglike molecules.
 | ||
| Fig. 9 UMAP visualization of the chemical space spanned by DataWarrior, iBonD aqueous, and the IUPAC dataset presented herein. UMAP was performed on the 2048-bit radius-3 count-based Morgan fingerprints computed for each chemical species. The dashed circles indicate regions of chemical space where most of the data are from this IUPAC dataset (bottom-right large cluster, topmost small cluster) or have few IUPAC data (the others). | ||
Fig. 10 shows that the distributions of the molecular weights covered by the IUPAC, iBonD, and DataWarrior sets are very similar, focusing on small organic compounds, though iBonD includes more large molecules. The great majority of the molecules in all 3 datasets have molecular weights of less than 300 amu, which may limit the applicability of these datasets for modeling larger, more complex biochemical or pharmaceutical compounds.
 | ||
| Fig. 10 Comparison of molar weights of datasets in this study. The histograms nearly completely overlap, indicating that the IUPAC, iBonD, and DataWarrior data have a very similar molecular weight coverage – centered on small molecules. The iBonD data includes slightly more large molecules than the other two datasets. | ||
The distributions of data are shown in Fig. 11. Compared to iBonD, the IUPAC dataset includes more pKaH values and a stronger left tail (very weak bases). Like the others, it includes peaks at approximately 4 and 9 pKa units, and a weak right tail. These provide further evidence that the experimental datasets span a generally similar set of compounds.
 | ||
| Fig. 11 Comparison of the pKa value distributions between the IUPAC, iBonD, and DataWarrior datasets. Overlapping bins are shown in purple. | ||
Usage in machine learning
Another use case we foresee is usage of the data in machine learning models. Large quantities of high-quality data are important for developing accurate predictive models.Before further discussion of this dataset, we must first comment on some highly-pervasive misusage of pKa data, which has thus far evaded discussion in the literature. After addressing these issues, we then discuss the modeling results.
Evaluation datasets
pKa models are generally evaluated on two common test sets, described below.Data leakage in existing datasets
To test the predictive power of a model, the test data should not overlap with training data. Data leakage, wherein the same molecule appears in both the test and training data, makes models appear more accurate than they really are. The ChEMBL, iBonD, and DataWarrior datasets, along with this dataset, all include some degree of overlap with the SAMPL and Novartis test sets, which could cause data leakage.Four compounds in the Novartis acid and base datasets27 are also present in this dataset. Among those four, three have exact matches for pKa type; one compound is in the dataset for a different acidity type (perrin3511) and may not be considered a leak.
All of these potential data leaks, from all datasets, are presented in the Zenodo repository associated with this manuscript. As both this dataset and others may be used in machine learning efforts, future works should be careful to remove these data from the training sets if evaluating on SAMPL or Novartis data to avoid data leakage.
The SAMPL7 challenge data also include two macroscopic pKa measurements that correspond to an inexact quantity; e.g. “pKa >12.” It is not clear whether these values were pruned, or assumed to be equal to 12 when performing tests using SAMPL7. We urge researchers who use those datasets to clearly state how those values were processed.
Finally, we note that these data are for macroscopic pKa values. In order to convert these into microscopic pKa, the modeler must compute the energetics of each relevant microstate.32,33 However, this process can be computationally expensive. If the modeler is using macroscopic data to predict values pertaining to a specific acidity center, then the modeler is actually reporting a microscopic pKa, and assuming their numerical values are identical (which is true for monoprotic acids and bases, but there can be substantial differences for polyprotic species). If this is done, the modeler should clearly state this assumption.
Extracting only pKa1 and pKaH1 values with high confidence near room temperature, we trained 10 separate Chemprop models, striping the training, validation, and test data in an 80%/10%/10% split such that all data in the whole data corpus was in a test split (the IUPAC model in Fig. 12). We also trained a set of 10 models on the ChEMBL dataset (labeled ChEMBL). Finally, we created a set of ten models (labeled Finetuned), wherein each model was initialized from a ChEMBL model and then finetuned on a split of the IUPAC data. That is, for each split i from 1 to 10, a model was trained on split i of ChEMBL and finetuned on split i of IUPAC. We pruned the training and pre-training data to avoid any overlaps with the test data in both the ChEMBL and IUPAC sets, and the ChEMBL data were further pruned to remove overlapping IUPAC compounds.
 | ||
| Fig. 12 Tukey's Honestly Significant Difference test applied to all test predictions, shown for the IUPAC, ChEMBL, and finetuned models, all trained using Chemprop v2.2. Each label includes 10 constituent models trained on separate training splits; these form the bounded ranges shown in the plots. Each dot corresponds to an average of all 10 model predictions. Test splits come from the IUPAC aqueous digitized data; and from the SAMPL and Novartis datasets, which largely include druglike molecules. Blue represents the “best”; gray shows statistically equivalent; and red shows statistically worse performance. | ||
We examined three test cases: (1) the held-out test data from the IUPAC dataset, mostly representing a large variety of small molecules; (2) the SAMPL dataset, representing a few classes of druglike fragments; (3) the Novartis dataset, representing a broader diversity of druglike molecules.
In total, this yields 3 sets of models (IUPAC, ChEMBL, Finetuned), differing only in the data used to train them as well as their hyperparameters; and 3 test sets (IUPAC splits, SAMPL, Novartis). We conducted Tukey's Honestly Significant Difference (HSD) test66 as implemented in SciPy,67,68 to better assess whether statistically significant differences exist between the models.
Fig. 12 shows the performances of the models along with the corresponding Tukey's HSD tests. Each model consists of ten substituent models trained on separate splits of the data. For each composite model under examination (IUPAC, ChEMBL, or Finetuned), the ten substituent models produce the distribution of RMSEs illustrated in Fig. 12.
In all cases, the model pretrained on ChEMBL data and then finetuned on IUPAC data performed the best or statistically identically to the best models. For the small molecules data, the IUPAC-based models perform the best, but on the other hand, the ChEMBL model did much better than the baseline IUPAC model on the SAMPL and Novartis test sets. Only the finetuned model appears to have the most versatile test performance, with good performance across both small molecules and druglike molecules.
The errors shown here are slightly higher than those of other recently-released models. However, we emphasize that care was taken to remove molecules that appear in the test splits from the training splits.
All the models mentioned above, including our recommended best model created from the 10 finetuned models, are available for download; see the Data Availability section. We recommend leveraging the whole ensemble of the Finetuned model to make predictions; this is readily done in the Chemprop software, which can use all 10 finetuned models to obtain an average prediction and estimate ensemble uncertainty (or leverage the other suite of uncertainty tools available).
Potential limitations of dataset
We anticipate that several outstanding challenges may require additional effort for some computational workflows. We encourage readers to report such errors when they are discovered so that they may be fixed in future versions of the dataset.We sometimes observed that different translation algorithms returned different SMILES strings, often due to inconsistencies in how locants are parsed. In these cases, as well as when zero or one algorithm(s) returned a valid result, we manually constructed SMILES strings based on the provided names, following IUPAC naming rules. Errors in SMILES strings may appear due to either failures in machine translation, inaccuracies in manual curation, or errors in the source material, though we expect such errors to be infrequent.
Another challenge is providing representative microstructures at the corresponding experimental conditions. Many of the compounds, including amino acids, are predominantly zwitterionic at room temperature and neutral pH conditions, which has implications for the pKa values as well. However, we herein identify those species by their neutral tautomers only, as these are easier to obtain and common to search. Also, since these are generally macroscopic pKa data, the structural distinctions are not crucial to using the data productively. We hence made no effort to represent any additional potentially relevant tautomers for each compound; each compound is labeled with just one SMILES string (and thereby one isomeric form). The SMILES strings provided in the dataset correspond to the tautomers provided by the SMILES translation software, or in the case of manual translation, by whichever form most literally was provided by the IUPAC name. Therefore, amino acids and all other potentially zwitterionic compounds are represented in their uncharged form.
Additionally, the “pKa types” of some amphoteric molecules were manually assigned. Several pK values were sometimes provided in the reference works without any indication of dissociation type (e.g. pKa versus pKaH). Although we only included pKa types we could assign with high confidence, it is still possible that some errors were made during this transcription phase.
The original tabulation of the source pKa data was not always done in a consistent fashion, including the designation of acidity types and orderings. For instance, an entry may have been provided with pK1 and pK2 values, but the values may correspond to pKaH1 and pKa1, or to two acidic dissociations, or to two pKaH values. The authors of this work used their best judgment and cross-referenced experimental data to other literature and predictions by ChemAxon's Protonation software, but it is still possible that this step has introduced additional errors, especially in the Serjeant compilation. Indeed, the Serjeant compilation recommends citing the Perrin sources as the “major source of pK values” for amphiprotic compounds, which may include duplicated entries in the acidic and basic compilations. During data validation, we checked for compounds with large discrepancies, and manually corrected those. However, there is the possibility that we missed subtler errors or mislabelings.
Some data may be duplicated between the Perrin and Serjeant compilations, as both compilations were conducted independently. We include all available sets of compounds here without combining entries that appear in both compilations.
It is possible that inconsistencies or errors exist in the original tabulation, or differences exist among the three text compilations that may contradict one another. We urge the reader to check the associated references for desired datapoints when relying on individual datapoints.
Finally, as mentioned earlier, the chemical coverage of the experimental data may not be conducive toward certain applications, e.g. for modeling large pharmaceutical compounds. We anticipate that incorporating computed thermodynamic information with the data, or explicitly partitioning pKa into group-based contributions, will help improve the generalizability of the predictive models.
We intend for this dataset to be a “living” resource, in the sense that any errors discovered throughout its usage can be corrected.
Conclusion
We have provided a digitized dataset that can be used for reference by researchers or as a data source for data-driven models. The dataset includes metadata such as temperature, references, method of measurement, assessed reliability, pressure, and general comments. We have also commented on and demonstrated use of the data for modeling applications, pointing out numerous cases where data leakage is present in commonly-used datasets for pKa prediction. We emphasize once more that these data are macroscopic pKa values, and therefore models trained on this data will only predict macroscopic pKa.We hope that this work joins other collections of pKa data as a reference for applications in organic chemistry and computational modeling. We emphasize again that this digitization may include some errors, which can be corrected in future versions of the dataset. Because references are supplied, users can cross-reference a tabulated value with its source publication.
Finally, we hope that this work, in conjunction with high-throughput experimentation and advancements in data mining that sample diverse regimes of chemical space, will lead to the development of even more capable open-source tools and libraries for pKa prediction used broadly in chemistry-related applications.
Author contributions
All authors contributed to the development and writing of the manuscript. J. W. Z. conducted the database generation, analysis, and model development. O. L.-J. conducted additional data cleaning and analysis. W. H. G. managed and planned the work.Conflicts of interest
There are no conflicts to declare.Data availability
The latest version of the IUPAC dataset is publicly available at: https://doi.org/10.5281/zenodo.7236452. At present, the data is provided under a CC BY-NC 4.0 license.The models and data splits used in this study are available at: https://doi.org/10.5281/zenodo.18165948. The Chemprop package can be used to run the models, and is freely available on GitHub at https://github.com/chemprop/chemprop/.
Supplementary information (SI): pdf including additional examples of data analysis, information about reference works, model training details, and data pruning. See DOI: https://doi.org/10.1039/d6ra02418a.
Acknowledgements
J. W. Z. and W. H. G. acknowledge the Machine Learning for Pharmaceutical Discovery and Synthesis Consortium (MLPDS) for funding. J. W. Z. further acknowledges funding from a Takeda Fellowship. We gratefully acknowledge Ye Li, Leah McEwen, and Stuart Chalk for many helpful discussions and invaluable contributions toward facilitating this work. We also acknowledge IUPAC members David Shaw, Glenn Hefter, and executive director Dr Lynn Soby. We acknowledge Prof. Ivo Leito, Sofja Tshepelevitsh, and Ivari Kaljurand for thoughtful discussions. We further acknowledge members of the Green group who assisted with data checking, including: Yunsie Chung, Xiaorui Dong, Michael Forsuelo, Kevin Greenman, Esther Heid, and Hao-Wei Pang. We thank PubChem scientists including Evan Bolton and Jeff Zhang for their support in resolving SMILES strings from IUPAC names, and for integration within PubChem. We thank the Internet Archive for providing the digitized copy of Perrin (1965) with IUPAC's permission. We further acknowledge usage of Amazon Textract and ChemAxon Molconverter (https://chemaxon.com). We thank Prof. Markus Kraft for many thoughtful discussions regarding this work.References
- D. T. Manallack, R. J. Prankerd, E. Yuriev, T. I. Oprea and D. K. Chalmers, The significance of acid/base properties in drug discovery, Chem. Soc. Rev., 2013, 42, 485–496 RSC.
- A. Sabljic and Y. Nakagawa, Non-First Order Degradation and Time-dependent Sorption of Organic Chemicals in Soil, ACS Publications, 2014, pp 85–118 Search PubMed.
- L. Mamy, D. Patureau, E. Barriuso, C. Bedos, F. Bessac, X. Louchart, F. Martin-Laurent, C. Miege and P. Benoit, Prediction of the fate of organic compounds in the environment from their molecular properties: A review, Crit. Rev. Environ. Sci. Technol., 2015, 45, 1277–1377 CrossRef CAS PubMed.
- R. P. Bell, The Proton in Chemistry, Springer Science & Business Media, 2013 Search PubMed.
- R. Lu, J. Sun, Y. Wang and Z. He, Quantitative structure-retention relationship studies with biopartitioning micellar chromatography systems by amended linear solvation energy relationships in consideration of electronic factor, Chromatographia, 2009, 70, 21–29 CrossRef CAS.
- I.-H. Um, H.-J. Han, J.-A. Ahn, S. Kang and E. Buncel, Reinterpretation of curved hammett plots in reaction of nucleophiles with aryl benzoates: change in rate-determining step or mechanism versus ground-state stabilization, J. Org. Chem., 2002, 67, 8475–8480 CrossRef CAS PubMed.
- I.-H. Um, J.-Y. Lee, H.-T. Kim and S.-K. Bae, Curved Hammett plot in alkaline hydrolysis of O-aryl thionobenzoates: change in rate-determining step versus ground-state stabilization, J. Org. Chem., 2004, 69, 2436–2441 CrossRef CAS PubMed.
- E. A. Castro, R. Aguayo, J. Bessolo and J. G. Santos, Kinetics and mechanism of the reactions of S-2, 4-dinitrophenyl 4-substituted thiobenzoates with secondary alicyclic amines, J. Org. Chem., 2005, 70, 7788–7791 CrossRef CAS PubMed.
- J. W. Zheng and W. H. Green, Experimental Compilation and Computation of Hydration Free Energies for Ionic Solutes, J. Phys. Chem., 2023, 127, 10268–10281 CrossRef CAS.
- L. C. Kröger, S. Müller, I. Smirnova and K. Leonhard, Prediction of solvation free energies of ionic solutes in neutral solvents, J. Phys. Chem., 2020, 124, 4171–4181 CrossRef PubMed.
- T. Nevolianis, J. Zheng, S. Müller, M. Baumann, S. Tshepelevitsh, I. Kaljurand, I. Leito, I. Smirnova, W. Green and K. Leonhard, Solvation free energies of anions: from curated reference data to predictive models, J. Am. Chem. Soc., 2025, 147, 30626–30646 CrossRef CAS PubMed.
- M. Nic, L. Hovorka, J. Jirat, B. Kosata and J. Znamenacek, IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 2005 Search PubMed.
- IUPAC Gold Book: Hydron, 2025, DOI:10.1351/goldbook.H02904.
- C. L. Perrin, I. Agranat, A. Bagno, S. E. Braslavsky, P. A. Fernandes, J.-F. Gal, G. C. Lloyd-Jones, H. Mayr, J. R. Murdoch and N. S. Nudelman, et al., Glossary of terms used in physical organic chemistry (IUPAC Recommendations 2021), Pure Appl. Chem., 2022, 94, 353–534 CrossRef CAS.
- P. Pracht, R. Wilcken, A. Udvarhelyi, S. Rodde and S. Grimme, High accuracy quantum-chemistry-based calculation and blind prediction of macroscopic pKa values in the context of the SAMPL6 challenge, J. Comput.-Aided Mol. Des., 2018, 32, 1139–1149 CrossRef CAS PubMed.
- M. Işık, A. S. Rustenburg, A. Rizzi, M. R. Gunner, D. L. Mobley and J. D. Chodera, Overview of the SAMPL6 p K a challenge: evaluating small molecule microscopic and macroscopic p K a predictions, J. Comput. Aided Mol. Des., 2021, 35, 131–166 CrossRef PubMed.
- S. Prasad, J. Huang, Q. Zeng and B. R. Brooks, An explicit-solvent hybrid QM and MM approach for predicting pKa of small molecules in SAMPL6 challenge, J. Comput. Aided Mol. Des., 2018, 32, 1191–1201 CrossRef CAS PubMed.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa Prediction for Organic Acids and Bases, Chapman & Hall, 1981 Search PubMed.
- F. H. Vermeire and W. H. Green, Transfer learning for solvation free energies: From quantum chemistry to experiments, Chem. Eng. J., 2021, 418, 129307 CrossRef CAS.
- Y. Chung, F. H. Vermeire, H. Wu, P. J. Walker, M. H. Abraham and W. H. Green, Group Contribution and Machine Learning Approaches to Predict Abraham Solute Parameters, Solvation Free Energy, and Solvation Enthalpy, J. Chem. Inf. Model., 2022, 62, 433–446 CrossRef CAS PubMed.
- F. H. Vermeire, Y. Chung and W. H. Green, Predicting Solubility Limits of Organic Solutes for a Wide Range of Solvents and Temperatures, J. Am. Chem. Soc., 2022, 144, 10785–10797 CrossRef CAS PubMed.
- Y. Chung and W. H. Green, Machine learning from quantum chemistry to predict experimental solvent effects on reaction rates, Chem. Sci., 2024, 2410–2424 RSC.
- S. Biswas, Y. Chung, J. Ramirez, H. Wu and W. H. Green, Predicting critical properties and acentric factors of fluids using multitask machine learning, J. Chem. Inf. Model., 2023, 63, 4574–4588 CrossRef CAS PubMed.
- K. A. Spiekermann, L. Pattanaik and W. H. Green, Fast predictions of reaction barrier heights: toward coupled-cluster accuracy, J. Phys. Chem., 2022, 126, 3976–3986 CrossRef CAS.
- K. P. Greenman, W. H. Green and R. Gómez-Bombarelli, Multi-fidelity prediction of molecular optical peaks with deep learning, Chem. Sci., 2022, 13, 1152–1162 RSC.
- X. Pan, H. Wang, C. Li, J. Z. Zhang and C. Ji, MolGpka: A Web Server for Small Molecule pKa Prediction Using a Graph-Convolutional Neural Network, J. Chem. Inf. Model., 2021, 61, 3159–3165 CrossRef CAS PubMed.
- M. Baltruschat and P. Czodrowski, Machine learning meets pKa, Chem. Inf. Sci., 2020, 9, 1–12 Search PubMed.
- J. Wu, Y. Wan, Z. Wu, S. Zhang, D. Cao, C.-Y. Hsieh and T. M. F. Hou, SuP.-pKa: Multi-fidelity modeling with subgraph pooling mechanism for pKa prediction, Acta Pharm. Sin. B, 2022, 2572–2584 Search PubMed.
- Q. Yang, Y. Li, J. D. Yang, Y. Liu, L. Zhang, S. Luo and J. P. Cheng, Holistic Prediction of the pKa in Diverse Solvents Based on a Machine-Learning Approach, Angew. Chem., Int. Ed., 2020, 59, 19282–19291 CrossRef CAS PubMed.
- J. Xiong, Z. Li, G. Wang, Z. Fu, F. Zhong, T. Xu, X. Liu, Z. Huang, X. Liu and K. Chen, et al., Multi-instance learning of graph neural networks for aqueous pKa prediction, Bioinformatics, 2022, 38, 792–798 CrossRef CAS PubMed.
- F. Mayr, M. Wieder, O. Wieder and T. Langer, Improving small molecule pKa prediction using transfer learning with graph neural networks, Front. Chem., 2022, 10, 866585 CrossRef CAS PubMed.
- W. Luo, G. Zhou, Z. Zhu, Y. Yuan, G. Ke, Z. Wei, Z. Gao and H. Zheng, Bridging Machine Learning and Thermodynamics for Accurate pKa Prediction, JACS Au, 2024, Search PubMed.
- C. Wagen, Physics-Informed Machine Learning Enables Rapid Macroscopic pKa Prediction, ChemRxiv, 2025, preprint, ChemRxiv:2025-t8s9z-v2, DOI:10.26434/chemrxiv-2025-t8s9z-v2.
- O. Abarbanel and G. Q. K. Hutchison, QupKake: Integrating Machine Learning and Quantum Chemistry for Micro-pKa Predictions, J. Chem. Theor. Comp., 2025, 20, 6946–6956 CrossRef PubMed.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich and B. Al-Lazikani, et al., ChEMBL: a large-scale bioactivity database for drug discovery, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- M. Davies, M. Nowotka, G. Papadatos, N. Dedman, A. Gaulton, F. Atkinson, L. Bellis and J. P. Overington, ChEMBL web services: streamlining access to drug discovery data and utilities, Nucleic Acids Res., 2015, 43, W612–W620 CrossRef CAS PubMed.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez and J. F. Mosquera, et al., The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods, Nucleic Acids Res., 2024, 52, D1180–D1192 CrossRef CAS PubMed.
- C. Yang, C. Gong, Z. Zhang, J. Fang, W. Li, G. Liu and Y. Tang, In Silico Prediction of pKa Values Using Explainable Deep Learning Methods, J. Pharm. Anal., 2024, 101174 Search PubMed.
- ChemAxon Marvin, https://chemaxon.com/marvin.
- J. W. Zheng, I. Leito and W. H. Green, Widespread Misinterpretation of pKa Terminology for Zwitterionic Compounds and Its Consequences, J. Chem. Inf. Model., 2024, 64, 8838–8847 CrossRef CAS PubMed.
- ChemAxon Docs: Red and blue representation of pKa values, https://docs.chemaxon.com/display/docs/calculators_red-and-blue-representation-of-pka-values.md, Accessed: 7-31-2024.
- J.-P. Cheng, J.-D. Yang, X.-S. Xue, P. Ji, X. Li and Z. Wang, iBonD Website. https://ibond.nankai.edu.cn/, Accessed: 01 September 2024 Search PubMed.
- H. An, X. Liu, W. Cai and X. Shao, AttenGpKa: a universal predictor of solvation acidity using graph neural network and molecular topology, J. Chem. Inf. Model., 2024, 64, 5480–5491 CrossRef CAS PubMed.
- T. Sander, J. Freyss, M. Von Korff and C. Rufener, DataWarrior: An open-source program for chemistry aware data visualization and analysis, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- S. Dimitrov, R. Diderich, T. Sobanski, T. Pavlov, G. Chankov, A. Chapkanov, Y. Karakolev, S. Temelkov, R. Vasilev and K. Gerova, et al., QSAR Toolbox–workflow and major functionalities, SAR QSAR Environ. Res., 2016, 27, 203–219 CrossRef CAS PubMed.
- I. Sushko, S. Novotarskyi, R. Körner, A. K. Pandey, M. Rupp, W. Teetz, S. Brandmaier, A. Abdelaziz, V. V. Prokopenko and V. Y. Tanchuk, et al., Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information, J. Comput. Aided Mol. Des., 2011, 25, 533–554 CrossRef CAS PubMed.
- R. Fraczkiewicz, M. Lobell, A. H. Goller, U. Krenz, R. Schoenneis, R. D. Clark and A. Hillisch, Best of both worlds: Combining pharma data and state of the art modeling technology to improve in silico pKa prediction, J. Chem. Inf. Model., 2015, 55, 389–397 CrossRef CAS PubMed.
- R. Fraczkiewicz, H. Quoc Nguyen, N. Wu, N. Kausch-Busies, S. Grimbs, K. Sommer, A. Ter Laak, J. Günther, B. Wagner and M. Reutlinger, Best of both worlds: An expansion of the state of the art pKa model with data from three industrial partners, Mol. Inf., 2024, 43, e202400088 CrossRef CAS PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, Chemprop: a machine learning package for chemical property prediction, J. Chem. Inf. Model., 2023, 64, 9–17 CrossRef PubMed.
- E. P. Serjeant and B. Dempsey, Ionisation constants of organic acids in aqueous solution, IUPAC Chem. Data Ser., 1979, 23, 160–190 Search PubMed.
- D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solutions, Franklin Book Company, 1965, vol. 1 Search PubMed.
- D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solutions: Supplement, Franklin Book Company, 1972, vol. 1 Search PubMed.
- A. M. Slater, The IUPAC aqueous and non-aqueous experimental pKa data repositories of organic acids and bases, J. Comput. Aided Mol. Des., 2014, 28, 1031–1034 CrossRef CAS PubMed.
- G. Kortüm, W. Vogel and K. Andrussow, Disssociation constants of organic acids in aqueous solution, Pure Appl. Chem., 1960, 1, 187–536 CrossRef.
- K. Izutsu, Acid-base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell Scientific Publications Oxford, 1990, vol. 35 Search PubMed.
- Amazon Textract, https://aws.amazon.com/textract/.
- D. M. Lowe, P. T. Corbett, P. Murray-Rust and R. C. Glen, Chemical name to structure: OPSIN, an open source solution, J. Chem. Inf. Model., 2011 Search PubMed.
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang and S. H. Bryant, PubChem: a public information system for analyzing bioactivities of small molecules, Nucleic Acids Res., 2009, 37, W623–W633 CrossRef CAS PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He and B. A. Shoemaker, et al., PubChem substance and compound databases, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen and B. Yu, et al., PubChem 2019 update: improved access to chemical data, Nucleic Acids Res., 2019, 47, D1102–D1109 CrossRef PubMed.
- Chemical Identifier Resolver, https://cactus.nci.nih.gov/chemical/structure, Accessed: 12-1-2025.
- S. Müller, How to crack a SMILES: automatic crosschecked chemical structure resolution across multiple services using MoleculeResolver, J. Cheminf., 2025, 17, 117 Search PubMed.
- R. Fraczkiewicz, What, This “Base” Is Not a Base? Common Misconceptions about Aqueous Ionization That May Hinder Drug Discovery and Development, J. Med. Chem., 2025 Search PubMed.
- L. McInnes, J. Healy and J. Melville, UMAP: Uniform manifold approximation and projection for dimension reduction, arXiv, 2018, preprint, arXiv:1802.03426, DOI:10.48550/arXiv.1802.03426.
- D. E. Graff, N. K. Morgan, J. W. Burns, A. C. Doner, B. Li, S.-C. Li, J. Manu, A. Menon, H.-W. Pang, H. Wu, A. S. Zalte, J.W. Zheng, C. W. Coley, W. H. Green and K. P. Greenman, Chemprop V2: an Efficient, Modular Machine Learning Package for Chemical Property Prediction, J. Chem. Inf. Model., 2025, 66, 28–33 CrossRef PubMed.
- J. W. Tukey, Comparing individual means in the analysis of variance, Biometrics, 1949, 99–114 CrossRef CAS.
- D. Chmiel, S. Wallan and M. Haberland, tukey_hsd: An accurate implementation of the Tukey honestly significant difference test in Python, J. Open Source Softw., 2022, 7, 4383 CrossRef.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser and J. Bright, et al., SciPy 1.0: fundamental algorithms for scientific computing in Python, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- I. Leito, I. Kaljurand, M. Piirsalu, S. Tshepelevitsh, J. W. Zheng, M. Rosés and J.-F. Gal, Acid dissociation constants in selected dipolar non-hydrogen-bond-donor solvents (IUPAC Technical Report), Pure Appl. Chem., 2025 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |